Biochemie und Pathobiochemie: Druckversion
Vorwort
BearbeitenDieses Buch ist all jenen gewidmet, die sich mit der Biochemie des menschlichen Körpers näher beschäftigen möchten. Dem Leser möge das Buch als „Reiseführer“ dienen, um sich im ebenso faszinierenden wie verworrenen Labyrinth der Stoffwechselwege zurecht zu finden.
Die Reihenfolge der Kapitel auf der Startseite ist angelehnt an die Übersicht der KEGG Pathway Database. Anhand einer Übersichtsseite kann man sich weiterhin die wichtigsten Verknüpfungspunkte der verschiedenen Wege klar machen. Anders als ein herkömmliches Lehrbuch bietet ein E-Book die Möglichkeit, die Vernetzung der Stoffwechselwege durch Querverweise (Links) so nachzubilden, dass man sich relativ mühelos durch das Netzwerk der metabolischen Pfade klicken kann. So kann der Leser die Zusammenhänge der verschiedenen Wege bequem im Detail explorieren, z.B. das Woher und Wohin der C1-Reste (Folatstoffwechsel) oder wo überall eine bestimmte Aminosäure eine Rolle spielt.
Die graphische Darstellung der einzelnen Wege erfolgt in einer weitgehend standardisierten Tabellen-Form, die einen Überblick geben und möglichst viele Informationen zusammenfassen soll. Man findet hier Angaben zu den Substraten und Reaktionspartnern, die umgesetzt werden, zu den beteiligten Cofaktoren, zu den die Reaktionen katalysierenden Enzymen inklusive ihrer Regulationsmöglichkeiten und assoziierten Erkrankungen (Enzymdefekte). Um das, was in den Stoffwechselwegen passiert, auch intuitiv begreifbarer zu machen sind neben den Hauptsubstraten auch wichtige Reaktionspartner farblich markiert, so z.B. die energiereichen Phosphate und Reduktionsäquivalente, die beide eine Form der Energiewährung in der Zelle darstellen. Weiterhin das energiearme Endprodukt sämtlicher Abbauwege Kohlendioxid bzw. Bicarbonat, das größtenteils abgeatmet wird oder wie andere C1-Reste dazu genutzt werden kann, um bestimmte Moleküle mit einzelnen Kohlenstoffatomen auszustatten. Und letztlich der Stickstoff, der z.B. im Aminosäuren- und Nukleotidstoffwechsel eine bedeutende Rolle spielt. Auch die Stellschrauben der Substratflüsse, die „Schlüsselenzyme“ sind derart hervorgehoben.
Die Begleittexte werden ergänzt durch Bezüge zu den klinischen Fächern (Pathologie, Pharmakologie/Toxikologie, Labordiagnostik u.a.), um auch hier das Verständnis der Zusammenhänge zu erleichtern. Die hereditären Stoffwechseldefekte (inborn errors of metabolism), mit denen jeder Arzt früher oder später in Berührung kommt, werden ausführlich in eigenen Kapiteln beleuchtet.
Ein großer Dank gilt Benutzer:NEUROtiker für das Zeichnen der zahllosen chemischen Formeln.
Und zu guter Letzt: Die Autoren freuen sich über Kritik, Anregungen und Verbesserungsvorschläge!
Die Bausteine des Lebens
BearbeitenDie Zusammensetzung des Körpers aus den Elementen
Bearbeiten| Das Periodensystem mit den für das Leben bedeutsamen Substanzen. | ||||||||||||||||||||||
| H | He | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Li | Be | B | C | N | O | F | Ne | |||||||||||||||
| Na | Mg | Al | Si | P | S | Cl | Ar | |||||||||||||||
| K | Ca | Sc | Ti | V | Cr | Mn | Fe | Co | Ni | Cu | Zn | Ga | Ge | As | Se | Br | Kr | |||||
| Rb | Sr | Y | Zr | Nb | Mo | Tc | Ru | Rh | Pd | Ag | Cd | In | Sn | Sb | Te | I | Xe | |||||
| Cs | Ba | La | * | Hf | Ta | W | Re | Os | Ir | Pt | Au | Hg | Tl | Pb | Bi | Po | At | Rn | ||||
| Fr | Ra | Ac | ** | Rf | Db | Sg | Bh | Hs | Mt | Ds | Rg | |||||||||||
| * | Ce | Pr | Nd | Pm | Sm | Eu | Gd | Tb | Dy | Ho | Er | Tm | Yb | Lu | ||||||||
| ** | Th | Pa | U | Np | Pu | Am | Cm | Bk | Cf | Es | Fm | Md | No | Lr | ||||||||
| ||||||||||||||||||||||
Die Grundelemente
BearbeitenVon der Antike bis weit ins Mittelalter nahm man an, dass der Körper aus den vier Elementen Erde, Wasser, Luft und Feuer besteht. Heute wissen wir, dass sich alle Lebewesen überwiegend aus den vier Elementen Sauerstoff (Oxygenium), Kohlenstoff (Carbonium), Wasserstoff (Hydrogenium) und Stickstoff (Nitrogenium) bzw. ihrer Verbindungen zusammensetzen. Diese Nichtmetalle bilden zusammen bereits etwa 95 % unseres Körpergewichts. Die vier Hauptgruppenelemente wurden von der Natur offensichtlich deswegen bevorzugt, weil sie stabile kovalente Bindungen und eine niedrige Masse bieten.
Sauerstoff (63 %) und Wasserstoff (10 %) liegen überwiegend in der Verbindung Wasser (H2O) vor, aus dem wir zu 2/3 bis 3/4 bestehen. Während wir das Wasser als äußeren Lebensraum vor etwa 400 Millionen Jahren verlassen haben, bevorzugen unsere Zellen nach wie vor ein wässriges Milieu für ihre Lebensäußerungen, weswegen sich Landbewohner das Wasser sozusagen mitgenommen haben. Wasser ist aufgrund seiner besonderen chemisch-physikalischen Eigenschaften als Trägerlösung sämtlicher biochemischer Reaktionen und als Transportmedium innerhalb und außerhalb der Zelle essentiell.
| Zusammensetzung des menschlichen Körpers.
Bezogen auf 70 kg Körpergewicht. Aus Flindt 1995, nach Heidermanns 1957, Kleiber 1967. | ||
| Element | Gew-% ca. | Masse ca. |
|---|---|---|
| Sauerstoff (O) | 63 | 44 kg |
| Kohlenstoff (C) | 20 | 14 kg |
| Wasserstoff (H) | 10 | 7 kg |
| Stickstoff (N) | 3 | 2,1 kg |
| Kalzium (Ca) | 1,5 | 1 kg |
| Phosphor (P) | 1 | 0,7 kg |
| Kalium (K) | 0,25 | 170 g |
| Schwefel (S) | 0,2 | 140 g |
| Chlor (CI) | 0,1 | 70 g |
| Natrium (Na) | 0,1 | 70 g |
| Magnesium (Mg) | 0,04 | 30 g |
| Eisen (Fe) | 0,004 | 3 g |
| Kupfer (Cu) | 0,0005 | 300 mg |
| Mangan (Mn) | 0,0002 | 100 mg |
| Iod (I) | 0,00004 | 30 mg |
So besitzt Wasser z.B. eine hohe Wärmekapazität, kann gut Protonen abgeben und aufnehmen und ist polar (Dipol), es besitzt eine optimale Viskosität und Oberflächenspannung sowie einen hohen Siede- und niedrigen Gefrierpunkt. Auch für die Physiologie (Wärmeregulation) und Ökologie (Dichteanomalie des Wassers) sind die besonderen Eigenschaften des Wasser bedeutsam, so dass die Existenz von Leben ohne die Existenz von Wasser schwer vorstellbar erscheint.
Sauerstoff (2 mögliche Bindungen) und besonders Wasserstoff (1 Bindung) kommt weiterhin in den meisten organischen Molekülen vor. Beide sind auch an der zellulären Atmung beteiligt. Ein Wasserstoffatom besteht aus einem Proton (H+) und einem Elektron (e-), daher werden Wasserstoffübertragungen auch zur Elektronenübertragung genutzt, um z.B. chemische Energie zu transferieren.
Stickstoff (3 Bindungen) findet sich besonders in Aminosäuren, den Bausteinen der Proteine und in Purinen und Pyrimidinen, den Bausteinen der DNA und RNA.
Kohlenstoff bildet das „Skelett“ aller organischen Moleküle. Seine Fähigkeit, gleichzeitig 4 Bindungen eingehen zu können (4 Außenelektronen), ist die Grundlage für die unglaubliche Vielfalt an einfachen und komplexen organischen Molekülen, die die Natur hervorbringt. Die freien „Ärmchen“ der Kohlenstoffgerüste werden im einfachsten Falle von Wasserstoff besetzt, weswegen man es sich bei den Strukturformeln meist spart, sie einzuzeichnen. Derartige Kohlenwasserstoffe sind unpolar und deswegen wasserabweisend (hydrophob). Durch Modifikation mit anderen Atomen, die sog. funktionelle Gruppen bilden (z.B. basische Aminogruppen (-NH2 bzw. -NH3+) oder saure Carboxylgruppen (-COOH bzw. -COO-)) sowie das Einfügen von Doppelbindungen und anderes mehr gewinnen die Moleküle ihre Vielfalt an charakteristischen chemischen Eigenschaften und Interaktionsmöglichkeiten.
Die Mengenelemente
BearbeitenNeben den 4 Grundelementen dominieren die 7 Mengenelemente das molekulare Leben, die ebenfalls zu den Hauptgruppenelementen gehören. Es handelt sich um die Alkalimetalle Natrium (Na) und Kalium (K), die Erdalkalimetalle Magnesium (Mg) und Kalzium (Ca), die Nichtmetalle Phosphor (P) und Schwefel (S) und das Halogen Chlor (Cl). Die Metalle geben ihre ein oder zwei äußeren Elektronen leicht ab, Halogene füllen sich die Lücke in der äußersten Elektronen-Schale gerne auf (Oktettregel). Daher liegen diese Elemente im wässrigen Milieu des Körpers meist in Form von Ionen (geladene Teilchen, Elektrolyte) vor: Na+, K+, Mg2+, Ca2+ und Cl–. Diese Ionen spielen eine große Rolle bei der Regulation des osmotischen Drucks und des Wasserhaushalts (bes. Natrium), für elektrische Aktivitäten (Ruhepotential und Aktionspotentiale an Muskel- und Nervenzellen, bes. Kalium und Natrium) und Transportvorgänge an Zellmembranen (Na+/K+-Pumpe, Na+/Glucose-Symporter u.a.) sowie als sekundärer Botenstoff in der Zelle (Kalzium z.B. in Muskelzellen) und Cofaktor der Blutgerinnung (Kalzium). Chlor findet sich auch im Magen in Form von Salzsäure (HCl = H+ + Cl–). Phosphor und Schwefel findet man häufig in Form der Säureanionen Phosphat (HPO42–) und Sulfat (SO42–).
Kalzium bildet zusammen mit Phosphat in Form von Kalzium-Hydroxylapatit (Ca5(PO4)3(OH)) den (extrazellulär abgelagerten) mineralischen Anteil des Knochens und der Zähne, was sich auch entsprechend in den Gewichtsprozenten eines Wirbeltiers (siehe Tabelle) niederschlägt. Phosphat findet sich weiterhin in vielen organischen Molekülen. Beispiele sind die Nukleotide wie z.B. Adenosintriphosphat (ATP), die für den Energiestoffwechsel wichtig sind (energiereiche Phosphorsäureanhydridbindungen) und aus denen die Nukleinsäuren (DNA und RNA) bestehen und die Phospholipide, die die Zellmembranen aufbauen (der polare hydrophile Phosphatkopf weist zur wässrigen Phase). Phosphate sind auch an der Regulation des Säure-Basen-Haushalts beteiligt (Phosphatpuffer).
Schwefel findet man z.B. in den Aminosäuren Cystein und Methionin, in den Vitaminen Thiamin (B1) und Biotin sowie in den Glycosaminoglycanen Keratansulfat, Chondroitinsulfat (Knorpel, Haare, Nägel) und Heparansulfat (gerinnungsaktiv). Weiterhin findet man sog. Eisen-Schwefel-Zentren in den Proteinkomplexen der Atmungskette.
Die Spurenelemente
BearbeitenSpurenelemente sind wie der Name schon sagt nur in Spuren im Körper vorhanden, trotzdem sind zumindest die essentiellen Spurenelemente lebensnotwendig. Viele davon gehören zu den Nebengruppenelementen bzw. Übergangsmetallen. Man findet sie vielfach als funktionstragende Elemente in Enzymen. Daneben sind sie wichtig für weitere Funktionen: Eisen ist z.B. das Zentralion des Häm-Moleküls, das man in verschiedenen Enzymen (z.B. Cytochrom P450) findet, aber auch als das Sauerstoff-Bindungsmolekül im Hämoglobin und Myoglobin. Jod ist ein Bestandteil der Schilddrüsenhormone. Zink stabilisiert die Speicherform des Insulins.
Die Biomoleküle
BearbeitenAus den oben beschriebenen Elementen rekrutieren sich neben einfachen anorganischen Verbindungen wie Wasser, Hydrogencarbonat und Kalziumphosphat (Kalzium-Hydroxylapatit) die organischen Moleküle aus denen alle Lebewesen aufgebaut sind. Vier große Gruppen nehmen hier eine besondere Stellung ein:
- Proteine: Alle Proteine sind aus einem Pool von etwa 20 Aminosäuren aufgebaut. Mit etwa 18 % haben Sie den größten Anteil an einer menschlichen Zelle direkt nach dem Wasser. Proteine erfüllen strukturelle Funktionen, etwa im Bindegewebe, in den Muskeln oder beim Zytoskelett. Außerdem sind sie in Form von Enzymen und Transportproteinen maßgeblich am Stoffwechsel und Transportprozessen beteiligt. Einige Aminosäuren und ihre Derivate erfüllen weitere Funktionen, z.B. als Signalmoleküle (Bsp.: Die Aminosäuren Glycin, Glutamat, Aspartat, die Schilddrüsenhormone L-Thyroxin und Trijodthyronin sowie die biogenen Amine Histamin und Serotonin).
- Lipide (Fette): Sie stellen durchschnittlich 5 % der Zellmasse und dienen einerseits zur Speicherung von Energie in Form der Di- und Triglyceriden in Fettzellen, andererseits bilden sie das Gerüst der Membranen, die sich in Zellen und um Zellen herum befinden, und ermöglichen so eine Kompartimentierung (Gliederung in chemische Reaktionsräume). Ferner gehören einige der Botenstoffe im Körper zur Gruppe der Lipide.
- Kohlenhydrate (Zucker): Ungefähr 2 % des Gewichts einer menschlichen Zelle fallen auf diese Stoffgruppe. Sie sind die primäre Energiequelle für den Körper, allen voran Glucose. Zur schnellen Verfügbarkeit werden auch sie in Energiespeichern in Form von Glykogen eingelagert, diese Speicher sind jedoch recht begrenzt. Weiterhin finden sich Kohlenhydrate als „Ketten“ oder „Bäume“ an Proteine gehaftet, dadurch ergänzen sie deren funktionelles Spektrum, vor allem im extrazellulären Raum und spielen eine Rolle bei der Zell-Zell-Interaktion.
- Nukleinsäuren: Die wohl wichtigste Aufgabe von Nukleinsäuren besteht in der Speicherung und Weitergabe der Erbinformation, vermittelt durch die DNA. Daneben spielen sie – in Form verschiedener Klassen von RNA – auch eine essentielle Rolle in der Umsetzung dieser Information in den Aminosäuren-Code der Proteine. In menschlichen Zellen sind sie zu durchschnittlich 1,5 % der Masse vertreten.
Mit Ausnahme der Lipide liegen alle diese Stoffgruppen als Polymere vor oder können zu solchen verknüpft werden. Die Natur erreicht dadurch, dass aus wenigen, einfachen, monomeren Bausteinen durch Kombination eine riesige Anzahl komplexer Moleküle aufgebaut werden kann, die an die jeweiligen Anforderungen angepasst sind.
| Monomere | Polymere |
|---|---|
| Aminosäuren | Oligopeptide und Proteine (Eiweiße) |
| Monosaccharide (z.B. Glucose, Fruktose, Galaktose) | Disaccharide (z.B. Saccharose, Lactose) und Polysaccharide (z.B. Stärke, Glycogen, Heteroglycane). |
| Purin- und Pyrimidin-Nukleotide | Nukleinsäuren (z.B. Desoxyribonukleinsäure (DNA) und Ribonukleinsäure (RNA)) |
Diese Mono- und Polymere haben charakteristische Eigenschaften:
- Polarität – Sowohl die Monomere als auch die Polymere sind meist nicht symmetrisch, sondern haben zwei verschiedene Enden. Proteine haben ein N-terminales und ein C-terminales Ende, Kohlenhydrate haben oft ein reduzierendes und ein nicht-reduzierendes Ende, Nukleinsäuren haben pro Einzelstrang ein 3'-OH- und ein 5'-OH-Ende.
- Informationsgehalt – Proteine/Peptide/Aminosäuren, Kohlenhydrate, Lipide u.a. Moleküle können Informationen tragen, die in Form der 3-dimensionalen Struktur kodiert ist. Die Decodierung erfolgt durch ein Molekül, das zu dieser Struktur komplementär aufgebaut ist. So erkennt z.B. der Insulinrezeptor spezifisch das Insulin-Peptid, ein Immunglobulin (Antikörper) erkennt spezifisch sein Antigen (z.B. ein Virus) und ein Enzym erkennt spezifisch die Substrate, die es umsetzt. In der DNA und RNA werden Informationen hingegen in einer abstrakten oder „digitalen“ Form verschlüsselt. So wird z.B. die Information für die Aminosäurensequenz eines Proteins auf der DNA – ein sog. Gen – in Form eines 4-Buchstaben-Codes abgelegt, wobei jede Aminosäure von 3 Buchstaben, d.h. einem Basentriplett, kodiert wird.
Neben den beschriebenen vier großen Gruppen kommen noch weitere chemische Verbindungen im Körper vor, wie anorganische Ionen (z.B. Phosphat oder Hydrogencarbonat), sowie eine Vielzahl organischer Verbindungen (z.B. Harnstoff, viele Vitamine), die sich nicht in eine dieser Gruppen einteilen lassen. Sie machen zusammen etwa 3,5 % der Masse einer Zelle aus.
Weblinks
BearbeitenEnergetik chemischer Reaktionen
BearbeitenEinführung
BearbeitenIm folgenden Abschnitt geht es um einige Grundprinzipien, die dem Ablauf chemischer Reaktionen zugrundeliegen. Auch wenn man die Formeln anfangs vielleicht nicht ganz versteht, so kann man sich doch vielleicht mit Hilfe der Alltagserfahrung eine Vorstellung davon machen, was passiert, wenn eine Zelle große Moleküle in kleinere zerlegt oder umgekehrt große aus kleinen aufbaut und wann sie dabei Energie gewinnt oder aufbieten muss.
Wenn beispielsweise Holz im Kamin verbrennt und der Umgebung Wärme spendet, dann passiert dabei folgendes: Die im Brennholz enthaltenen großen Zellulose-Moleküle werden bei der Reaktion mit Sauerstoff in eine viel größere Anzahl einfacher energiearmer Bausteine (Kohlendioxid, Wasser) zerlegt und die dabei frei werdende Energie wird in Form von Wärme an die Umgebung abgegeben. Dass dieser Prozess freiwillig abläuft, und zwar hier nur in eine bestimmte Richtung, hat einen bestimmten Grund: Das Holz strebt wie alle Systeme einen energiearmen Zustand an. Dafür muss es Energie z.B. in Form von Wärme an die Umgebung abgeben. Diese Energie steckt z.B. in den chemischen Bindungen der komplexeren Moleküle wie der Zellulose. Sie steckt aber auch in der „Ordnung“ der Struktur. Dass Systeme zur Unordnung neigen und nur unter Einsatz von Energie zu beherrschen sind kennt jeder, der einen Haushalt führt oder einen Garten pflegt. Dass insgesamt trotzdem soviel Ordnung auf unserem Planeten zu finden ist verdanken wir primär der Sonne, die uns ständig Energie in Form von Licht und Wärme zuführt. Die Lichtenergie wird vor allem von grünen Pflanzen in chemische Energie umgewandelt, in Form von Glucose, Zellulose u.a. energiereichen Moleküle gespeichert und über die Nahrungskreisläufe weiterverteilt.
Thermodynamik
BearbeitenHauptsätze
BearbeitenDie zwei Hauptsätze der Thermodynamik lauten:
1. Hauptsatz: Die gesamte Energie in einem System und seiner Umgebung bleibt konstant.
2. Hauptsatz: Bei spontan ablaufenden Vorgängen kann die Entropie (die „Unordnung“) eines Systems und seiner Umgebung nie kleiner werden.
Reaktionsenthalpie H
BearbeitenAngenommen wird eine Reaktion der Form A + B ⇔ C + D (A und B reagieren miteinander zu C und D).
A und B sind hier die Edukte, C und D die Produkte. Die Reaktion kann prinzipiell in beide Richtungen ablaufen, Hin- und Rückreaktionen laufen gleichzeitig ab.
Die Änderung der inneren Energie, der sog. Reaktionsenthalpie ΔH zwischen Produkten und Edukten einer Reaktion entspricht unter isobaren und isothermen Bedingungen der Summe aus der Reaktionswärme und der geleisteten Arbeit. Die Arbeit ist hier vernachlässigbar, so dass wir die Änderung der Reaktionsenthalpie ΔH mit der Reaktionswärme gleichsetzen können.
- Ist ΔH < 0, dann ist die Reaktion exotherm, gibt also Energie in Form von Wärme an die Umgebung ab.
- Ist ΔH > 0, so ist die Reaktion endotherm, d.h. sie nimmt Energie in Form von Wärme aus der Umgebung auf.
Reaktionsentropie S
BearbeitenAuch die Entropie ist eine Form der Energie in einem System. Die Entropie ist ein Maß für die Unordnung eines Systems. Mit wachsender Unordnung wächst die Entropie und die Energie nimmt ab. Umgekehrt muss Energie aufgewendet werden, um in einem System Ordnung zu erzeugen und die Entropie zu vermindern.
Gibbs freie Energie
BearbeitenFür Gibbs freie Energie (= freie Reaktionsenthalpie) gilt: ΔG = ΔH – T ∙ ΔS
- Ist ΔG < 0, so ist die Reaktion exergon, d.h. sie kann Arbeit leisten und sie läuft freiwillig ab.
- Ist ΔG > 0, so ist die Reaktion endergon und sie läuft nur dann ab, wenn Arbeit investiert wird.
ΔG ist ein Maß für die Änderung der Gesamtenergie im System und damit für die Triebkraft der Reaktion. Die Triebkraft nimmt zu (d.h. ΔG ist umso negativer), je positiver die Entropieänderung ΔS und je negativer die Reaktionsenthalpie ΔH. D.h. in einfachen Worten: Eine Reaktion läuft umso freiwilliger ab, je mehr dabei die Unordnung zunimmt und je mehr Wärmeenergie dabei frei wird. Mit zunehmender Temperatur T in Kelvin steigt der Einfluss der Entropieänderung ΔS.
Chemische Gleichgewichte
BearbeitenWenn das chemische Gleichgewicht zwischen Edukten und Produkten erreicht ist, dann gilt: ΔG = 0 und die Hinreaktion läuft genauso schnell ab wie die Rückreaktion. Die aktuelle Triebkraft ΔG hängt von den aktuellen Konzentrationen K der beteiligten Edukte und Produkte ab.
| ΔG = ΔG0 + R ∙ T ∙ ln K | mit K = [C]∙[D]/[A]∙[B] |
ΔG0 ist die Energieänderung, die notwendig ist oder frei wird, wenn die Edukte vollständig in die Produkte überführt werden. Dieser Wert ist damit ein Maß für die Lage des Gleichgewichts. Ist ΔG0 positiv, die Reaktion als endergon, so liegt das Gleichgewicht eher links bei den Edukten, ist ΔG0 negativ, die Reaktion als exergon, so liegt das Gleichgewicht eher rechts bei den Produkten.
Für die Gleichgewichtslage mit ΔG = O ergibt sich aus der Formel:
ΔG0 = – R ∙ T ∙ ln KGW
D.h. ΔG0 ist temperaturabhängig und lässt sich aus den Konzentrationen der Reaktionspartner bestimmen, wenn sich das chemische Gleichgewicht eingestellt hat (R ist die allgemeine Gaskonstante).
Gekoppelte Reaktionen
Bearbeiten
Endergone Reaktionen (ΔG0 positiv) laufen auch dann effektiv ab, wenn sie mit exergonen gekoppelt werden. Bsp.:
| Reaktion 1 | ||
| Reaktion 2 | ||
| Gesamtreaktion: |






Das ΔG0 Ges der Gesamtreaktion ergibt sich aus der Summe von ΔG0 1 und ΔG0 2. In biologischen Systemen werden energetisch ungünstige Reaktionen (z.B. Biosynthesen) häufig mit der Hydrolyse von ATP (ATP ⇒ ADP + Pi) gekoppelt, eine stark exergone Reaktion. Die Energie steckt dabei vorwiegend in der Phosphorsäureanhydrid-Bindung zwischen dem zweiten und dritten Phosphat-Rest des ATPs. Ein zweiter wichtiger Energiezwischenspeicher ist reduziertes Nicotinamid-adenin-dinucleotid(-phosphat), abgekürzt: NAD(P)H + H+. Hier liegt die Energie in Form der Reduktionskraft vor, d.h. in der Fähigkeit energiereiche Elektronen zu übertragen. Während NADPH/H+, das z.B. im Pentosephosphatweg regeneriert wird, Biosynthesen antreiben kann, speist NADH/H+ seine Elektronen überwiegend in die Atmungskette ein zur ATP-Gewinnung.
Im Körper kann so die Energie, die aus dem schrittweisen Abbau (Katabolismus) von Glucose, Aminosäuren und Fetten stammt und in Form von ATP und NADPH zwischengespeichert wurde, zur (endergonen) Biosynthese (Anabolismus) der verschiedensten Biomoleküle und anderer Prozesse (z.B. Transportvorgänge oder Bewegung) verwendet werden.
Fließgleichgewicht
BearbeitenDie vorgenannten Betrachtungen beziehen sich auf geschlossene Systeme, in denen sich nach einer gewissen Zeit ein stabiles Gleichgewicht einstellt.
Offene Systeme wie Lebewesen tauschen mit ihrer Umwelt jedoch ständig Energie und Stoffe aus. Dadurch kommt es bei den zahllosen chemischen Reaktionen im Körper praktisch nie zur Einstellung der Gleichgewichtslage, sondern die Edukte werden ständig in ihre Produkte umgewandelt. Dadurch bleiben auch die Intermediatkonzentrationen in einem gewissen Toleranzbereich weitgehend konstant.
Alle Lebewesen befinden sich mit ihrer Umgebung im Fließgleichgewicht, der sog. Homöostase. Sie sind auf ständige Energiezufuhr angewiesen, die auf unserem Planeten letztendlich von der Sonne stammt (E = H ∙ ν) und von den Pflanzen mit Hilfe der Photosynthese und Carbonfixierung chemisch fixiert wird.
Weblinks
Bearbeiten- Thermodynamics Review. The Medical Biochemistry Page.
Enzyme
BearbeitenAllgemeines
Bearbeiten
Enzyme katalysieren chemische Reaktionen, indem sie die Aktivierungsenergie herabsetzen. D.h. sie stabilisieren den energetisch ungünstigen Übergangszustand. Erst so können die chemischen Reaktionen auch bei Körpertemperatur in der notwendigen Geschwindigkeit ablaufen. Die endgültige Gleichgewichtslage bleibt davon jedoch unbeeinflusst, denn die hängt nur von der freien Reaktionsenthalpie (ΔG) ab.
Die katalysierte Reaktion kann prinzipiell in beide Richtung ablaufen. Weitere Eigenschaften sind die meist hohe Substratspezifität (Schlüssel-Schloss-Prinzip) und Reaktionsspezifität der Enzyme.
Enzyme werden nach den Reaktionen, die sie katalysieren, klassifiziert:
- Oxidoreduktasen katalysieren Redoxreaktionen.
- Transferasen übertragen funktionelle Gruppen von einem Substrat auf ein anderes.
- Hydrolasen spalten Bindungen unter Aufnahme von Wasser.
- Lyasen (Synthasen) spalten komplexere Produkte zu bzw. bilden sie aus einfacheren Substraten ohne Spaltung von ATP.
- Isomerasen katalysieren isomerische Umwandlungen.
- Ligasen (Synthetasen) spalten komplexere Produkte zu bzw. bilden sie aus einfacheren Substraten mit Spaltung von ATP.
Manche Enzyme zeigen eine größere Bandbreite an katalysierten Reaktionen und können mehreren Enzymklassen zugeordnet werden.
Mechanismen der enzymatischen Katalyse
Bearbeiten- Säure-Basen-Katalyse
- Kovalente Katalyse
- Metallionen-Katalyse
Cofaktoren
Bearbeiten- Metallionen
- Magnesium (Mg) - Magnesium wird typischerweise von Kinasen genutzt (Mg-ATP als Substrat).
- Eisen (Fe) - Häm-gebundenes Eisen ist an Elektronentransportvorgängen (Enzyme der mitochondrialen Atmungskette) sowie an verschiedenen Redoxreaktionen beteiligt (Cytochrom P450, Cyclooxygenase).
- Kupfer (Cu) - Bestandteil der zytoplasmatischen Kupfer,Zink-Superoxiddismutase (SOD).
- Zink (Zn) - Bestandteil der zytoplasmatischen Kupfer,Zink-Superoxiddismutase (SOD), der Carboanhydrase, der Carboxypeptidase, der alkalischen Phosphatase und von Steroidhormon-Rezeptoren.
- Mangan (Mn) - Bestandteil der mitochondrialen Mangan-Superoxiddismutase (SOD)
- Organische Verbindungen wie z.B. Vitamine, Biopterin.
Michaelis-Menten-Kinetik
Bearbeiten
Die Geschwindigkeit v des Substratumsatzes steigt zuerst linear mit der Substratkonzentration [S] und flacht dann zunehmend ab, bis das Enzym gesättigt und die Maximalgeschwindigkeit vmax erreicht ist. vmax hängt dann nur noch von der Enzymkonzentration [E] und der Wechselzahl des Enzyms kcat ab. Die Substratkonzentration in mmol/l, bei der das Enzym mit halbmaximaler Geschwindigkeit läuft nennt man Michaelis-Konstante Km. Je niedriger die Km, desto effizienter arbeitet das Enzym. Graphisch ergibt sich eine hyperbolische Sättigungskurve.
| mit Vmax = kcat x [E] |
![{\displaystyle v={V_{max}\times [S] \over K_{m}+[S]}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/930ae4b532a3858fc08e7c3dc0bfa5597e7a4a6f)
Lineweaver-Burk-Auftragung
Bearbeiten
Die Konstanten der Michaelis-Menten-Kinetik können durch doppelt reziproke Auftragung von Reaktionsgeschwindigkeit 1/V und Substratkonzentration 1/[S] ermittelt werden. Diese Darstellung wird Lineweaver-Burke-Diagramm genannt und ergibt eine Gerade. vmax kann aus dem y-Achsenabschnitt und Km aus der Steigung der Geradengleichung berechnet oder aus dem x-Achsenabschnitt abgelesen werden.
- y = Steigung mal x + y-Achsenabschnitt
![{\displaystyle {\frac {1}{V}}={\frac {K_{m}+[S]}{V_{max}\times [S]}}={\frac {K_{m}}{V_{\max }}}\cdot {\frac {1}{[S]}}+{\frac {1}{V_{\max }}}}](https://wikimedia.org/api/rest_v1/media/math/render/svg/7214ee9511b68400bb7eadcca115ca5c82e704a8)
Kooperativität
Bearbeiten
Manche Enzyme zeigen eine sigmoide Sättigungskurve. Beschrieben wurde diese Kinetik erstmals beim Sauerstoffbindungsverhalten des Hämoglobins. Ein Hämoglobin-Molekül besteht aus 4 Untereinheiten. Hat eine dieser Untereinheiten Sauerstoff gebunden, so ändert sich die räumliche Konformation und die restlichen Untereinheiten binden Sauerstoff leichter. Bezogen auf ein Enzym mit mehreren Bindungsstellen heißt das, dass die Affinität eines Enzyms für ein Substrat im Falle einer positiven Kooperativität mit der Zahl der besetzten Substrat-Bindungsstellen zunimmt.
Allosterie
BearbeitenEin allosterisch regulierbares Enzym besitzt zusätzlich zu der/den Substratbindungsstelle(n) weitere Bindungsstellen für andere Effektoren, die die Enzymaktivität modulieren können. Ein Beispiel dafür ist die Phosphofructokinase (Glycolyse), die durch den allosterischen Regulator Fructose-2,6-bisphosphat aktiviert wird.
Allosterische Liganden vom V-Typ senken die vmax. Allosterische Liganden vom K-Typ beeinflussen die Substratkonzentration, bei der 1/2 vmax erreicht wird, d.h. die Km.
Enzyminhibition
Bearbeiten
- Kompetitive Hemmung - Der dem Substrat strukturell ähnelnde Inhibitor konkurriert mit dem Substrat am aktiven Zentrum des Enzyms. Die Kinetik-Kurve wird nach rechts verschoben und die Km wird größer. Eine höhere Substratkonzentration kann die Hemmung durchbrechen.
- Nicht-kompetitive reversible Hemmung - Der Inhibitor bindet außerhalb des aktiven Zentrums am Enzym oder am Enzym-Substrat-Komplex. Gesenkt wird v.a. die vmax.
- Nicht-kompetitive irreversible Hemmung - Der Inhibitor bindet irreversibel und blockiert das Enzym. Die Hemmung ist mit einer höheren Substratkonzentration nicht zu durchbrechen.
- Suizidinhibitoren - Suizidinhibitoren werden im aktiven Zentrum des Enzym umgesetzt, lösen sich dann aber nicht von diesem ab. Das Enzym ist dauerhaft blockiert.
-
 a) normale Umsetzung
a) normale Umsetzung
b) kompetitive Hemmung -
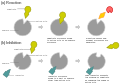 a) normale Umsetzung
a) normale Umsetzung
b) nicht-kompetitive Hemmung


Weitere Einflüsse auf die Enzymaktivität
Bearbeiten- Temperatur (Faustregel: Verdopplung der Geschwindigkeit bei einer Temperaturerhöhung um 10°C).
- pH - Enzyme haben bei einem bestimmten pH ihr Aktivitätsmaximum.
- Redoxgleichgewicht - Oxidations- und Reduktionsmittel können die Enzymaktivität beeinflussen.
Regulation im Organismus
BearbeitenKurzfristig:
- Änderungen der Substrat- und Produktkonzentrationen
- Allosterische Regulation z.B. in Form einer Aktivierung durch die Edukte oder als Rückkopplungshemmung (Feedback-Hemmung) durch die Produkte. Oder durch eigens gebildete Zwischenprodukte.
Mittelfristig:
- Interkonversion - Reversible kovalente Modifikation. Bsp.: Phosphorylierung des Enzyms durch spezifische Kinasen und Dephosphorylierung durch spezifische Phosphatasen.
- Lokalisation des Enzyms und Kompartimentierung der Enzymaktivitäten.
- Regulation der Halbwertszeit der mRNA (Translation) oder des Proteins (Enzym-Abbau durch Ubiquitinilierung).
- Limitierte Proteolyse - Aktivierung durch Abspaltung von bestimmten Peptidstücken am Pro-Enzym (Zymogen). Bsp.: Das Pankreasenzym Trypsinogen wird erst im Darm durch die Enteropeptidase in das aktive eiweißspaltende Trypsin umgewandelt.
Längerfristig:
- Regulation auf Ebene der DNA (Genexpression) - Repression oder Induktion der Enzyme durch Trankriptionsfaktoren.
Da Stoffwechselwege meist in Form von längeren Reaktionsketten, -kaskaden oder -zyklen organisiert sind können komplexe Stoffwechselprozesse effektiv über einzelnen Enzyme gesteuert werden, die häufig am Anfang des Stoffwechselwegs liegen und die Kaskade starten. Diese „Schrittmacherenzyme“ werden meist allosterisch oder durch Interkonversion reguliert.
Kohlenhydratstoffwechsel
BearbeitenBedeutung der Kohlenhydrate im Organismus
Bearbeiten- Wichtigster schnellverfügbarer Energielieferant, insbesondere für Glucose-abhängige Organe wie das Gehirn.
- Homopolysaccharide dienen Pflanzen (Stärke) und Tieren (Glycogen) als mittelfristige Energiereserve.
- Oligosaccharide sind ein wichtiger Bestandteil von Glycoproteinen und wichtig als Erkennungsstrukturen für den intrazellulären Transport (Protein-Targeting) und auf der Zelloberfläche (Bsp.: ABO-Antigene).
- Wichtiger Bestandteil der Heteropolysaccharide, die eine hohe Wasserbindungsfähigkeit aufweisen (OH-Gruppen) und die die Interzellularsubstanz aufbauen.
Stellung des Kohlenhydratstoffwechsels im Gesamtstoffwechsel
BearbeitenDie Glycolyse und der Citratzyklus bilden das Rückgrat des gesamten Stoffwechsels. Die Verbindungen zu den anderen Stoffwechselwegen werden in den einzelnen Kapiteln näher beleuchtet und sind zusammengefasst im Kapitel Glycolyse aufgeführt. Eine Übersicht über die verschiedenen Monosaccharide, die im Stoffwechsel eine Rolle spielen findet man hier.
Die Leber ist das Zentrum des Kohlenhydratstoffwechsels
BearbeitenDie Leber ist das wichtigste Zentrum des Glucosestoffwechsels und als „Nährstoff-Puffer“ zwischen Darm (Glucoseaufnahme nach dem Essen) und restlichen Kreislauf geschaltet. Die Leber speichert Glucose in Form von Glycogen und gibt die Glucose bei Bedarf wieder ab. Weiterhin ist sie zur Gluconeogenese fähig, d.h. zur Neubildung von Glucose z.B. aus Aminosäuren oder Lactat. Reguliert wird die Beschreitung der entgegengesetzten Wege über den Glucosetransporter GLUT2, die Glucokinase und Insulin einerseits, die die Glucoseaufnahme fördern und Glucagon, Katecholamine und Glucokortikoide andererseits, die die Glucoseabgabe (Energiebereitstellung für den Körper) begünstigen.
Bei Glucosezufuhr:
- gelangt Glucose über den Glucosetransporter 2 (GLUT2) vermehrt in die Leberzelle und aktiviert dort die Glucokinase (Enkopplung vom GkRP). Die meisten Glucosetransporter ermöglichen die Glucoseaufnahme durch erleichterte Diffusion (Uniport) entlang des Konzentrationsgradienten. GLUT2 wird exprimiert in der Leber, in den Pankreas-β-Zellen, in der apikalen Membran der Dünndarmmukosa und in der Niere. Er besitzt eine hohe Michaelis-Konstante von ca. 40 mmol/l (Km von GLUT1: ca. 20 mmol/l, von GLUT3: ca. 10 mmol/l), und transportiert Glucose nahezu konzentrationsabhängig im Rahmen der normalen Blutglucosespiegel.
- Insulin induziert die Transkription der Glucokinase (Hexokinase IV), das erste Schrittmacher-Enzym der Glycolyse und reprimiert die Glucose-6-phosphatase, das letzte Enzym der Gluconeogenese.
- Die Glucokinase (Hexokinase IV) (exprimiert in Leber- und Pankreas-B-Zelle) besitzt ebenfalls eine höhere Km von 8 mmol/l = 144 mg/dl, die etwa im Rahmen der Glucosespiegel im Portalblut liegt, als andere Hexokinasen (Km < 1 mmol/l, d.h. hohe Affinität und konzentrationsunabhängige Aufnahme) und arbeitet damit wie GLUT2 konzentrationsabhängig. Die konzentrationsabhängige Arbeitsweise von GLUT2 und Glucokinase sorgt dafür, dass sich die Glucoseaufnahme in die Leberzelle automatisch der Glucosemenge anpasst, die aus dem Verdauungstrakt anflutet.
- Ein weiterer Schalter, der durch Insulin umgelegt wird (und vice versa durch Katecholamine und Glucagon) ist die Synthesesteigerung des allosterischen Regulators Fructose-2,6-bisphosphat, der die Phosphofructokinase (Glycolyse) aktiviert und die Fructose-1,6-bisphosphatase (Gluconeogenese) hemmt.
- Das durch die Glucokinase vermehrt gebildete Glucose-6-phosphat (das im Ggs. zur Glucose die Zelle nicht mehr verlassen kann) hemmt die Glycogenolyse und fließt nun vermehrt in die Glycogensynthese, in die Glycolyse, den Hexosemonophosphatweg und die Saccharidsynthese.
Bei Nahrungskarenz, niedrigem Blutzucker und vermehrtem Bedarf (Sympathikusaktivierung) erhöhen Glucagon, Katecholamine (Adrenalin, Noradrenalin) und Glucokortikoide (Kortisol) den Blutzuckerspiegel durch Förderung der Gluconeogenese und Glycogenolyse v.a. in der Leber.
Die B-Zellen des Pankreas produzieren Insulin
Bearbeiten |
Das Pankreas ist eine exokrine (Verdauungsenzyme) und endokrine Drüse (Inselzellen: A-Zellen: Glucagon, B-Zellen: Insulin, D-Zellen: Somatostatin).
Wie die Leber exprimieren auch die B-Zellen der Bauchspeicheldrüse den GLUT2 und die Glucokinase. GLUT2 und die Glucokinase bilden zusammen einen Glucosesensor, der in den Pankreasinselzellen die Insulinsekretion steuert.
Mit steigendem Blutglucosespiegel nehmen Glucoseaufnahme und -abbau proportional zu und es wird vermehrt ATP gebildet. ATP schließt K+-Kanäle und depolarisiert dadurch die Zelle. Die Depolariation führt zur Öffnung von Ca2+-Kanälen. Der sekundäre Botenstoff Kalzium stimuliert daraufhin die Produktion und Freisetzung von Insulin.
Insulin steigert die Aufnahme von Glucose und dessen Weiterverstoffwechselung in Skelettmuskel (Glycogen-Bildung) und Fettgewebe (Biosynthese von Fettsäuren und Triglyceriden), sowie die Weiterverstoffwechselung von Glucose in der Leber (u.a. Bildung von Glycogen). Außerdem erhöht Insulin die Kalium-Aufnahme in die Zelle.
Pathobiochemie: Insulinmangel (Zerstörung der B-Zellen) und periphere Insulin-Resistenz (Abnahme der Rezeptorempfindlichkeit) führen zum Diabetes mellitus. Beim Typ I-Diabetes werden die B-Zellen durch einen Autoimmunprozess oft schon in der Kindheit zerstört. Beim Typ II-Diabetes, der meist im höheren Alter auftritt, steht die Insulinresistenz im Vordergrund, so dass die Insulinspiegel anfangs häufig sogar erhöht sind, im Verlauf die B-Zellen jedoch „ausbrennen“. Die Insulinresistenz wird bei genetischer Veranlagung gefördert durch Übergewicht und Bewegungsmangel. Durch die Zunahme von Übergewicht auch schon bei jungen Menschen erkranken zunehmend auch schon Kinder und Jugendliche am Typ II-Diabetes. Die erhöhten Glucosespiegel führen zur vermehrten Glycosylierung von Proteinen und darüber langfristig zu Gefäß- und Nervenschäden. Niereninsuffizienz, Retinopathie, Polyneuropathie und periphere Durchblutungsstörungen (diabetischer Fuß) sind die Spätfolgen. Akute Komplikationen sind die diabetische Ketoazidose (v.a. bei Typ I-Diabetes) durch intrazellulären Energiemangel und Enthemmung der Ketonkörpersynthese sowie das hyperosmolare Koma (v.a. bei Typ II-Diabetes), bei dem die steigenden Blutzuckerspiegel zu osmotischen Störungen führen (Polyurie mit Austrocknung: Ab einem BZ von 180 mg/dl kann die glomerulär frei filtrierte Glucose nicht mehr vollständig tubulär zurückresorbiert werden und es kommt zur Glucosurie mit osmotischer Diurese).
Insulin reguliert den Kohlenhydratstoffwechsel von Fett- und Muskelgewebe
Bearbeiten
In extrahepatischen Geweben ist Insulin der Schlüssel zur Glucoseaufnahme. Insulin ist das einzige Hormon, das den Glucose-Spiegel senken kann.
- Insulin fördert die Translokation von GLUT4 (Adipozyten, Muskelzellen) aus intrazellulären Vesikelmembranen in die Plasmamembran, so dass Fettgewebe und Skelettmuskel Glucose vermehrt aufnehmen.
- Insulin stimuliert weiterhin die Glycogensynthese (Muskel), die Glycolyse und die Fettsäuresynthese (Adipozyten).
Im Muskel kann die überschüssige Glucose als Glycogen gepeichert werden, im Fettgewebe kann sie in der Glycolyse zu Acetyl-CoA abgebaut und zur Lipidbiosynthese genutzt werden. Das im Muskel gespeicherte Glycogen wird bei Bedarf wieder in Glucose-6-phosphat umgewandelt, das aber nicht ins Blut übertreten kann (fehlende Glucose-6-phosphatase und daher keine Umwandlung in Glucose) und daher nur für den Eigenbedarf genutzt wird. Fettzellen stellen dem Körper bei Nahrungskarenz Fettsäuren und Glycerin zur Energiegewinnung zur Verfügung.
Das Gehirn ist auf ständige Glucosezufuhr angewiesen
BearbeitenDa das Gehirn ständig aktiv ist und normalerweise nur Glucose verwerten kann (in längerdauernden Hungerzeiten zunehmend auch Ketonkörper), ist es auf eine ständige und relativ konstante Glucosezufuhr angewiesen. Die Glucoseaufnahme erfolgt daher Insulin-unabhängig.
Weblinks
Bearbeiten
Stoffwechsel und Stoffwechselwege
Bearbeiten
Der Stoffwechsel
BearbeitenDer Stoffwechsel bzw. Metabolismus (μεταβολισμός, metabolismós (griech.): Stoffwechsel) ist die Gesamtheit aller (bio)chemischen Reaktionen, die in einem Organismus ablaufen. Er ist in Stoffwechselwege gegliedert, die in komplexer Weise zusammenhängen. Nach der Zielrichtung kann man den Stoffwechsel in einen Anabolismus (Aufbau, Biosynthesen) und einen Katabolismus (Abbau zur Energieerzeugung oder Gewinnung von Bausteinen für andere Biosynthesen) unterteilen. Beispielsweise werden im Verdauungstrakt die großen Moleküle der Nahrung wie Proteine, Kohlenhydrate (z.B. Stärke) und Fette in kleinere Einheiten - Aminosäuren, Einfachzucker und Fettsäuren - zerlegt und aufgenommen. Im Körper können diese dann weiter zerkleinert und zur Energiegewinnung in die Glycolyse und/oder den Citratzyklus eingeschleust werden oder der Körper baut sich daraus wieder eigene Substanz in Form von Proteinen, Kohlenhydraten (z.B. Glycogen) oder Fetten auf. Zur besseren Kontrolle sind anabole und katabole Wege innerhalb der Zelle häufig räumlich voneinander getrennt (Kompartimentierung), so findet man beispielsweise die β-Oxidation der Fettsäuren in der mitochondrialen Matrix, die Fettsäurenbiosynthese jedoch im Zytosol. Teilen sich zwei gegenläufige Stoffwechselwege bestimmte Intermediate/Enzyme im gleichen Kompartiment, so kann eine Regulationsmöglichkeit z.B. dadurch realisiert werden, indem einige Reaktionen in modifizierter Form mit unterschiedlicher Gleichgewichtslage ablaufen, die auch auch von jeweils eigenen Enzymen katalysiert werden. So sind z.B. 3 der 10 Reaktionen der Glycolyse (Glucose-Abbau) in der Gluconeogenese (Glucose-Bildung) durch 4 andere Reaktionen ersetzt, die das Gleichgewicht in die anabole Richtung verschieben. Einige Stoffwechselwege haben sowohl katabole als auch anabole Eigenschaften, man bezeichnet sie als amphibol. Klassisches Beispiel ist der Citratzyklus. Die zentrale Drehscheibe des Stoffwechsels oxidiert einerseits C2-Körper (katabol), andererseits nimmt sie Kohlenstoff-Körper mit 4 bis 6 C-Atomen aus verschiedenen Stoffwechselwegen auf und liefert sie in andere katabole (Pyruvatbildung -> Acetyl-CoA -> Oxidation) und anabole Wege (z.B. Gluconeogenese oder Häm-Synthese).
Vergleicht man anabole und katabole Wege, so stellt man fest, dass Abbauprozesse meist einen oxidativen Charakter haben (den Molekülen werden Elektronen entzogen), Aufprozesse hingegen eher reduktiv sind (Elektronen werden zugeführt). Mit den Elektronen wird letztlich eine Form von chemischer Energie transferriert. Wichtige Elektronen-Carrier im Stoffwechsel sind z.B. NAD+ und NAD(P)+. Eine weitere wichtige und universale Energiewährung ist ATP. ATP liefert Energie für anabole Prozesse und zerfällt dabei in ADP und anorganisches Phosphat. Die Energie zur Regeneration von ATP aus ADP und Pi in der Atmungskette stammt ebenfalls aus den Elektronen, die durch die o.g. oxidativen Abbauprozesse gewonnen werden. Daraus ergibt sich die zentrale Bedeutung von Redoxreaktionen und dem Redoxgleichgewicht für den Stoff- und Energiehaushalt der Zelle.
Die Regulation des Stoffwechsels erfolgt auf verschiedenen Ebenen und auf vielfältige Weise über Hormone, Stoffwechselintermediate u.a.m., die dann u. a. die Enzyme beeinflussen, die wie Wasserschleusen den Substratfluss durch das Labyrinth der Stoffwechselwege regeln.
Der Intermediärstoffwechsel, der im Mittelpunkt dieses Buches steht ist der bereits skizzierte Stoffwechsel der kleineren organischen Moleküle. Dieser umfasst die Prozesse des Lebens auf einer sehr basalen Ebene. Im Mittelpunkt des Interesses steht der chemische Auf-, Ab- und Umbau dieser Moleküle ineinander, ihre Funktionen und Eigenschaften sowie die Choreographie der Reaktionschritte und ihre Regulation.
Aus dem Intermediärstoffwechsel rekrutieren sich die Bausteine der „großen Moleküle“ wie Nukleinsäuren und Proteine (informations- und funktionstragende Biopolymere) bis hin zu den Zellmembranen und den verschiedenen Zellorganellen mit ihren spezifischen Aufgaben. Der letztgenannte Themenkomplex fällt im Allgemeinen unter die Begriffe Molekularbiologie und Zellbiologie und wird in einem eigenen Buch besprochen.
Stoffwechselwege
BearbeitenStoffwechselwege sind kaskadenartige Reaktionsketten, in denen ein bestimmtes Molekül auf-, ab- oder umgebaut wird. Sie können linear, divergierend/konfluierend oder zirkulär organisiert sein. Über Kurzschlüsse (gemeinsame Substrate) sind viele Stoffwechselwege miteinander zu einem komplexen Netzwerk verbunden. Die Endprodukte können gespeichert, direkt genutzt oder in anderen Wegen weiterverstoffwechselt werden.
Der erste Schritt einer Reaktionskette ist häufig irreversibel (das chemische Gleichgewicht liegt weit auf der Seite der Produkte) und wird meist durch ein Schrittmacherenzym kontrolliert. Die restlichen Schritte können sofern sie reversibel sind in beide Richtungen ablaufen, ungünstige und auch irreversible Reaktionsschritte können unter Einsatz von Energie (ATP, GTP, UTP, CTP, NAD(P)H/H+) mit günstigeren Reaktionen umgangen werden. Auf diese Weise können manche Stoffwechselwege je nach Bedarf in beide Richtungen ablaufen. Das einfachste Beispiel ist der zweite Teil des Pentosephosphatweges, bei dem die Flussrichtung nur von den Substratzu- und abflüssen abhängt. Weitere prominente Beispiele sind die Glycolyse (Glucose-Abbau) / Gluconeogenese (Glucose-Neubildung), die Glycogenbiosynthese und -degradation oder die schon sehr unterschiedlichen Abläufe der Fettsäurenbiosynthese und Fettsäurenoxidation.
Die Feinabstimmung der Substratflüsse erfolgt z.B. durch Feedback-Hemmung durch die Produkte oder bei zirkulären Reaktionen dadurch, dass jedes Produkt gleichzeitig das Edukt des nächsten Schrittes ist.
Koordination des Gesamtstoffwechsels (die „Stoffwechsellage“)
BearbeitenAnabolismus und Katabolismus des Gesamtorganismus werden vom vegetativen Nervensystem in Abhängigkeit von äußeren und inneren Faktoren eingestellt. Bei Aktivität (Kampf, Flucht, Hunger, Krankheit) dominiert der sympathische Teil des Nervensystems. Auf der Ebene des Stoffwechsels sorgt er u.a. dafür, dass dem Körper genug Energieträger (z.B. Glucose, Glycerin und Fettsäuren) für die Leistungserbringung zur Verfügung gestellt werden. Bei Ruhe, Nahrungsaufnahme, Ausscheidung, Wachstum und Regeneration dominiert der parasympathische Teil des Nervensystems und forciert die anabole Stoffwechsellage. An der Regulation des komplexen Wechselspiels von Aktivität und Erholung sind zahlreiche Hormone, Rezeptoren und Signaltransduktionskaskaden beteiligt (siehe dazu in den Büchern der Physiologie). Da am Ende vereinfacht gesagt jedoch nur zwei Konsequenzen stehen - anabole oder katabole Stoffwechsellage - erfolgt eine Integration der Informationen, indem viele Transduktionswege auf einer gemeinsamen Endstrecke münden, z.B. auf der Aktivierung oder Deaktivierung von Proteinkinasen, die wiederum bestimmte Schlüsselenzyme phosphorylieren oder dephosphorylieren.
Unter sympathischem Einfluss bei leerem Magen (vermittelt von Adrenalin, Noradrenalin, Glucagon) kommt es z.B. in der Leber und im Fettgewebe zu einer Hemmung der anabolen und Aktivierung der katabolen Wege* durch eine verstärkte Phosphorylierung folgender Enzyme:
- Acetyl-CoA-Carboxylase - Inaktivierung -> Hemmung der Fettsäurensynthese
- Glykogen-Synthase - Inaktivierung -> Hemmung der Glycogensynthese
- Pyruvat-Kinase - Inaktivierung -> Hemmung der Glycolyse
- Pyruvat-Dehydrogenase - Inaktivierung -> Hemmung der Acetyl-CoA-Bildung
- Hormon-sensitive Lipase - Aktivierung -> Verstärkte Lipolyse (Freisetzung von Glycerin und Fettsäuren)
- Glycogen-Phosphorylase - Aktivierung -> Verstärkter Glycogen-Abbau
- Fru-6-P-2-Kinase - Inaktivierung -> Hemmung der Glycolyse, Aktivierung der Gluconeogenese
Durch einen gemeinsamen Schalter (hier die Phosphorylierung) wird hier also der komplette Stoffwechsel auf Energiebereitstellung z.B. für die Muskeln umgeschaltet.
Der entgegengesetzte Effekt wird vom Wachstumshormon Insulin vermittelt. Insulin beendet über mehrere Zwischenschritte die Wirkung der Proteinkinasen und führt zur Dephosphorylierung. In der Folge kommt es zur Auffüllung der Reserven (Fett- und Glycogenbiosynthese) mit Senkung des Blutzuckerspiegels.
* Anm.: Katabol und anabol ist hier im Gesamtkontext (Stoffwechsellage) zu sehen. Glycolyse und Acetyl-CoA-Bildung sind für sich genommen zwar katabol, im oben beschriebenen Kontext liefern sie jedoch Substrat und Energie z.B. für die anabole Fettsynthese. Umgekehrt ist die Gluconeogenese aus z.B. Aminosäuren zwar anabol, geht aber hier zu Lasten der Körpersubstanz (Proteolyse).
Evolution
BearbeitenDie grundlegenden Stoffwechselwege wie Glycolyse, Citratzyklus, Fettsäurenauf- und -abbau, Nukleotidstoffwechsel und Hämbiosynthese sind ausgesprochen alt. Sie finden sich in allen drei Domänen der Lebewesen in sehr ähnlicher Form und waren schon lange am arbeiten, als sich vor über 1,5 Milliarden Jahren die entwicklungsgeschichtlichen Wege von Bakterien, Archaeen und Eukaryonten (Amöben, Tiere, Pflanzen) trennten. Die Konservierung dieser Wege kann auch als Hinweis dafür gelten, dass die Evolution als „Problemlösungsprozess“ zu diesem Zeitpunkt bereits die energetisch günstigsten Reaktionsfolgen (wenig Reaktionsschritte, hohe Effizienz/Energieausbeute) selektioniert hat, mit denen Lebewesen ihren Bau- und Energiestoffwechsel bestreiten müssen.
Umgekehrt haben viele Lebewesen bestimmte Stoffwechselwege aufgegeben, was unter energetischen Gesichtspunkten vorteilhaft ist, solange die nicht mehr selbstgebildeten Stoffe ausreichend verfügbar sind. So muss der Mensch z.B. (altersabhängig) 8 bis 10 der 20 proteinogenen Aminosäuren, bestimmte mehrfach ungesättigte Fettsäuren und die meisten Vitamine mit der Nahrung aufnehmen.
Literatur:
- Smith E et al. “Universality in intermediary metabolism”. Proc Natl Acad Sci U S A, 101(36):13168-73, Sep 7 2004. PMID 15340153
- Ebenhöh O et al. “Evolutionary optimization of metabolic pathways. Theoretical reconstruction of the stoichiometry of ATP and NADH producing systems”. Bull Math Biol, 63(1):21-55, Jan 2001. PMID 11146883
- Meléndez-Hevia E et al. “The puzzle of the Krebs citric acid cycle: assembling the pieces of chemically feasible reactions, and opportunism in the design of metabolic pathways during evolution”. J Mol Evol, 43(3):293-303, Sep 1996. PMID 8703096
- Romano AH et al. “Evolution of carbohydrate metabolic pathways”. Res Microbiol, 147(6-7):448-55, Jul-Sep 1996. PMID 9084754
- Koch AL. “How did bacteria come to be?”. Adv Microb Physiol, 40:353-99, 1998. PMID 9889982
- Ouzounis C et al. “The emergence of major cellular processes in evolution”. FEBS Lett, 390(2):119-23, Jul 22 1996. PMID 8706840
- Schmidt S. “Metabolites: a helping hand for pathway evolution?”. Trends Biochem Sci, 28(6):336-41, Jun 2003. PMID 12826406
- Light S et al. “Network analysis of metabolic enzyme evolution in Escherichia coli”. BMC Bioinformatics, 5:15, Feb 18 2004. PMID 15113413
- Alves R et al. “Evolution of enzymes in metabolism: a network perspective”. J Mol Biol, 320(4):751-70, Jul 19 2002. PMID 12095253
- Spirin V et al. “A metabolic network in the evolutionary context: multiscale structure and modularity”. Proc Natl Acad Sci U S A, 103(23):8774-9, Jun 6 2006. PMID 16731630
- Lawrence JG. “Common themes in the genome strategies of pathogens”. Curr Opin Genet Dev, 15(6):584-8, Dec 2005. PMID 16188434
- Wernegreen JJ. “For better or worse: genomic consequences of intracellular mutualism and parasitism”. Curr Opin Genet Dev, 15(6):572-83, Dec 2005. PMID 16230003
- Pál C et al. “Chance and necessity in the evolution of minimal metabolic networks”. Nature, 440(7084):667-70, Mar 30 2006. PMID 16572170
Stoffwechsel
BearbeitenVereinfachte Übersicht über wichtige humane Stoffwechselwege
(Proteinogene Aminosäuren in grün, Intermediate von Glycolyse/Gluconeogenese und Citratzyklus in beige.)
Bedeutung
Bearbeiten
Wie der Darstellung oben leicht zu entnehmen ist bilden die Glycolyse (Gluconeogenese) und der Citratzyklus das Rückgrat des gesamten Stoffwechsels. Einerseits wird hier der Zucker zur Energiegewinnung (ATP-Produktion) oxidiert, den die Pflanzen mit Hilfe der Photosynthese und Carbonfixierung produzieren. Anderseits liefern Glycolyse und Citratzyklus die Rohstoffe für zahlreiche Stoffwechselwege und nehmen umgekehrt deren Endprodukte auf, wenn sie nicht anderweitig ausgeschieden werden. Aufgefüllt wird dieses komplexe System durch die Nahrung, insbesondere durch Zucker, Fette und Aminosäuren. Darunter müssen insbesondere die essentiellen Aminosäuren und Fettsäuren aufgenommen werden, die der menschliche Körper nicht (mehr) synthetisieren kann. In kleinen Mengen müssen auch Vitamine und Mineralien zugeführt werden, sowie in großen Mengen Wasser, das der Körper ständig verliert. Wasser ist die Trägerlösung des gesamten Stoffwechsels.
Knotenpunkte
BearbeitenFolgende Substrate stellen wichtige Knotenpunkte im Stoffwechsel dar:
- Glucose - Bildung von Fructose und Glucose-6-phosphat für die Glycolyse
- α-D-Glucose-6-phosphat - G6P ist ein Allrounder-Molekül. Es kann für die Biosynthese von Glycogen, Galactose, Glucuronsäure und Inositol verwendet werden. Weiterhin kann es in den Pentosephosphat-Shunt einfließen und natürlich über die Glycolyse abgebaut werden. G6P stammt aus der Glucose (Nahrung), dem Glycogenabbau, der Gluconeogenese (und den daran hängenden Wegen) oder aus der Galactose. Im Ggs. zur Glucose können Glucose-6-phosphat und alle anderen phosphorylierten Intermediate der Glycolyse die Zelle nicht mehr verlassen.
- β-D-Fructose-6-phosphat - Bildung und Abbau von D-Fructose-2,6-bisphosphat, Verbindung zum Mannose- und Fucose-Stoffwechsel, Bildung und Abbau der Aminozucker, Endprodukt des HMP-Wegs bzw. hier reverser Eintritt.
- D-Glycerinaldehyd-3-phosphat / Dihydroxyacetonphosphat - Diese zwei Isomere verbinden die Glycolyse / Gluconeogenese mit dem Stoffwechsel der Triglyceride und Phosphoglyceride. Außerdem münden hier (ebenfalls zum Teil über Glycerin) die Abbauprodukte der Fructose ein.
- 3-Phosphoglycerat - Aus 3-Phosphoglycerat können die Aminosäuren Serin und Glycin gebildet werden und aus Serin wieder Cystein.
- Oxalacetat - Oxalacetat verbindet die Gluconeogenese (und indirekt die Glycolyse), den Citratzyklus und den Aspartat-/Asparagin-Stoffwechsel miteinander.
- Pyruvat - Pyruvat ist das Endprodukt der Glycolyse. Aus ihm kann Alanin gebildet werden. Umgekehrt können die Abbauprodukte von Tryptophan und Alanin sowie Cholin, Threonin, Glycin, Serin und Cystein hier eingeschleust werden. Pyruvat kann auch durch Decarboxylierung von Malat (durch das Malat-Enzym) aus dem Citratzyklus bezogen werden.
- Acetyl-CoA - Acetyl-Coenzym A ist ein wichtiger Scheidepunkt zwischen anabolen und katabolen Wegen. Acetyl-CoA ist einerseits der „Brennstoff“ des Citratzyklus, andererseits der Ausgangspunkt für zahlreiche Biosynthesen, v.a. für Fettsäuren, Ketonkörper und Cholesterin. Die aktivierte Essigsäure stammt hauptsächlich aus dem Abbau von Glucose, Glycerin, Fettsäuren und ketogenen Aminosäuren (Threonin, Lysin, Phenylalanin, Tyrosin, Tryptophan, Leucin, Isoleucin) und indirekt aus den damit verknüpften Wegen.
- α-Ketoglutarat - Das Intermediat des Citratzyklus stellt die Verbindung her zur „Stickstoff-Drehscheibe“ L-Glutamat, die wiederum die Aminosäuren Glutamin, Prolin und die Harnstoffzyklussubstrate Ornithin und Arginin liefert bzw. umgekehrt in den Citratzyklus einschleust. Histidin wird ebenfalls zu Glutamat abgebaut.
- Succinyl-CoA - Der Abbau von ungeradzahligen Fettsäuren, Threonin, Methionin, Valin und Isoleucin liefert Propionyl-CoA. Dieses kann nach Carboxylierung und Isomerisierung zu Succinyl-CoA an dieser Stelle in den Citratzyklus eingeschleust werden. Succinyl-CoA ist der Ausgangspunkt der Häm-Biosynthese.
- Fumarat - Entsteht aus Aspartat durch Stickstoffübertragung auf andere Moleküle und beim Abbau von Phenylalanin und Tyrosin.
Weblinks
Bearbeiten
Glycolyse
BearbeitenAllgemeines
BearbeitenDie Glycolyse (Embden-Meyerhof-Parnas-Weg, E.M.P.) wurde 1929 von Gustav Embden, Otto Meyerhof und Jakub Parnas aufgeklärt. Der Stoffwechselweg ist der wichtigste Weg des Glucoseabbaus. Glucose wird darin unter Energiegewinn in zwei kleinere Bruchstücke zerlegt. Die Glycolyse nimmt im Stoffwechsel durch die Verknüpfung mit vielen anderen anabolen und katabolen Wegen eine zentrale Stellung ein.
Teil 1: Spaltung von Glucose in Glycerinaldehyd-3-phosphat und Dihydroxyacetonphosphat
Bearbeiten| Tr. | All. | ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||||
| + Insulin (HK2) | - G6P | ATP
ADP |
|
2.7.1.1 | Tr | HK1-Def. | ||||||||
| + Insulin | - GKRP | Glucokinase (Hexokinase IV) | 2.7.1.2 | MODY2, PNDM, HHF3 | ||||||||||
|
||||||||||||||
| 6-Phospho- gluconat |
|
5.3.1.9 | Iso | GPI-Def. | ||||||||||
|
||||||||||||||
|
+ Insulin |
+ ADP, AMP, F-2,6-BP |
ATP
ADP |
|
2.7.1.11 | Tr | GSD7 (Tarui) | ||||||||
|
||||||||||||||
| Zn |
|
4.1.2.13 | Ly | GSD12, Hered. Fructoseintoleranz | ||||||||||
|
|
5.3.1.1 | Iso | TPI1-Def. | ||||||||||




Im ersten Teil wird ein C6-Körper in zwei einander ähnliche C3-Körper gespalten. Dafür muss das Kohlenhydrat zuerst mit 2 Phosphat-Resten präpariert werden, wofür 2 ATP investiert werden.
Teil 2: Abbau von Glycerinaldehyd-3-phosphat zu Pyruvat
Bearbeiten| Tr. | Kov. | All. | ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||||
| Pi + NAD+ | Pi + NAD+ |
|
1.2.1.12 | Ox | |||||||||
|
|||||||||||||
| ADP
ATP |
ADP
ATP |
|
2.7.2.3 | Tr | PGK1-Def. | ||||||||
|
|||||||||||||
|
5.4.2.1 | Iso | GSD10 | ||||||||||
|
|||||||||||||
|
H2O |
H2O |
Mg |
|
4.2.1.11 | Ly | ENO1-Def., GSD13 | |||||||
|
|||||||||||||
| + Insulin - cAMP |
- Phosph. | + F1,6BP - Alanin, ATP |
ADP
ATP |
|
2.7.1.40 | Tr | PKLR-Def., ATP-Erh. in RBC | ||||||
|
|||||||||||||
| NADH/H+ | NADH/H+ | L-Lactat-Dehydrogenase | 1.1.1.27 | Ox | GSD11, LDHB-Def. | ||||||||
|
|||||||||||||






Im 2. Teil der Glycolyse werden insgesamt 4 ATP (aus 2 Glycerinaldehyd-3-phosphat) zurückgewonnen.
Die Glycolyse
BearbeitenDie Glycolyse ist die wichtigste Stoffwechselreaktionskette überhaupt. Sie findet im Zytosol statt und spaltet ein Glucosemolekül (C6) in zwei Pyruvat (C3) mit einem Nettogewinn von 2 ATP und 2 NADH/H+. Hefepilze und manche Bakterien setzen Pyruvat auch zu Ethanol um (alkoholische Gärung: Decarboxylierung von Pyruvat durch die Pyruvatdecarboxylase (EC 4.1.1.1) zu Acetaldehyd, dann NADH/H+-abhängige Reduktion zum Alkohol durch die Alkoholdehydrogenase). Der ebenfalls anaerobe Abbau zum Lactat durch die Lactatdehydrogenase entspricht der bakteriellen Milchsäuregärung z.B. durch Milchsäurebakterien. Anaerob bezieht sich hier auf die Tatsache, dass die Endprodukte Lactat oder Alkohol nicht weiter unter Sauerstoff-Verbrauch abgebaut werden, d.h. über Citratzyklus und Atmungskette.
Beim anaeroben Abbau bis zum Alkohol oder Lactat wird der vorher (in der Glycerinaldehyd-3-phosphat-Dehydrogenase-Reaktion) entzogene Wasserstoff, der als reduzierendes NADH/H+ zwischengespeichert wurde, wieder auf die Endprodukte übertragen. So wird NAD+ regeneriert und das wichtige Redoxgleichgewicht neutral gehalten. Würde die Glycolyse unter anaeroben Bedingungen (bei anaeroben Bakterien oder beim Sauerstoffdefizit im Muskel) auf Pyruvat enden, so würde die Glycolyse wegen NAD+-Mangel sehr schnell zum Stillstand kommen.
Beim in der menschlichen Zelle üblicheren Abbau bis zum Pyruvat (der ebenfalls keinen Sauerstoff benötigt) entstehen zusätzlich zu den 2 ATP noch 2 NADH/H+. Diese können für Reduktionen (Elektronenübertragungen) verwendet werden oder wie die anderen Reduktionsäquivalente z.B. aus dem Citratzyklus zur ATP-Bildung in der Atmungskette genutzt werden. Letzteres setzt jedoch ein ausreichendes Sauerstoffangebot voraus, da die Elektronen (bzw. der Wasserstoff, der sich aus je einem Elektron und einem Proton zusammensetzt) am Ende der Atmungskette auf Sauerstoff übertragen werden müssen.
Die Reaktionen im Detail
BearbeitenDie Glycolyse lässt sich in zwei Teile gliedern. Im ersten Teil wird Glucose in zwei ähnliche Moleküle gespalten und es müssen pro Glucosemolekül 2 ATP investiert werden. Ähnlichkeit ist deswegen sinnvoll, damit der weitere Abbau gemeinsam stattfinden kann und nicht zwei separate Abbauwege unterhalten werden müssen. Im zweiten Teil werden 4 ATP (und 2 NADH/H+) zurückgewonnen.
Im Einzelnen laufen folgende Reaktionen ab:
- Die intrazelluläre Glucosekonzentration steht mit der extrazellulären im Diffusionsgleichgewicht. Damit die Zelle Glucose im Zytosol anreichern kann wandelt sie es mit Hilfe einer Hexokinase unter ATP-Verbrauch in Glucose-6-phosphat (G6P) um. So kann neue Glucose nachströmen. G6P kann die Zelle auch nicht mehr verlassen, da kein Transportmechanismus für G6P vorhanden ist. So werden mit der Hexokinase-Reaktion zwei Fliegen mit einer Klappe geschlagen.
- G6P kann nun in verschiedene Stoffwechselwege fließen, z.B. in die Glycogen-Synthese (Leber, Muskel) oder in den HMP-Weg. Soll G6P jedoch abgebaut werden, dann muss nun die Spaltung in zwei ähnliche Teile vorbereitet werden. G6P wird daher von der G6P-Isomerase zu Fructose-6-phosphat (F6P) isomerisiert und noch einmal von der Phosphofructokinase 1 ATP-abhängig phosphoryliert. Fructose-1-6-bisphosphat (F-1,6-BP) ist entstanden.
- F-1,6-BP wird nun von der F-1,6-BP-Aldolase in Glycerinaldehyd-3-phosphat (GAP) und Dihydroxyacetonphosphat (DHAP) gespalten. Bei den zwei C3-Körpern handelt es sich um Konstitutionsisomere. Sie können von der Triosephosphat-Isomerase leicht ineinander überführt werden und damit gemeinsam weiterverstoffwechselt werden. Der erste Teil der Glycolyse ist damit abgeschlossen und es folgt die Pay-off-Phase, beginnend mit (zwei) GAP.
- Die Oxidation der Aldehydgruppe des GAP zur Carbonylgruppe mit Hilfe der GAP-Dehydrogenase liefert 1,3-Bisphosphoglycerat (BPG). Diese Reaktion ist ausgesprochen exergon und das wird voll genutzt. Erstens wird dabei ein Reduktionsäquivalent (NADH/H+) gewonnen, zweitens ist noch genug Energie übrig, um auch noch ein anorganisches Phosphat an die neu entstandene Carboxyl-Gruppe zu heften. Letzteres macht sich in der nächsten Reaktion bezahlt.
- Die Phosphoglycerat-Kinase überträgt die Phosphatgruppe vom BPG nun auf ADP, so dass ein ATP gewonnen wird. Dies nennt man Substratkettenphosphorylierung. 3-Phosphoglycerat (3PG) ist das Endprodukt.
- Nun ist noch eine zweite Phosphatgruppe vorhanden, die gewinnbringend eingesetzt werden kann. Dafür wird 3PG zu 2PG isomerisiert (Phosphoglycerat-Mutase) und Wasser abgespalten (Enolase). Dabei kommt Phosphoenolpyruvat (PEP) heraus. PEP kann aufgrund seines hohen Gruppenübertragungspotentials als nächstes in einer zweiten Substratkettenphosphorylierung sein Phosphat auf ADP übertragen und ein weiteres ATP erzeugen. Übrig bleibt Pyruvat, das verschiedentlich weiterverstoffwechselt werden kann oder das bei Sauerstoffdefizit zum Lactat reduziert wird.
Energiebilanz
BearbeitenEs werden 2 ATP investiert und über die Substratkettenphosphorylierung 4 zurückgewonnen. D.h. der anaerobe Glucoseabbau bis zum Pyruvat liefert 2 ATP. Beim Abbau bis zum Lactat bleiben zusätzlich noch 2 NADH/H+ übrig, die bei der vollständigen Oxidation über Citrazyklus und Atmungskette ca. 32 ATP liefern, also 16 mal mehr als der anaerobe Abbau der Glucose. Dafür setzt der aerobe Abbau aber auch ein ausreichendes Sauerstoffangebot und die entsprechenden Enzyme voraus, sowie beim Eukaryonten das Vorhandensein von Mitochondrien.
Regulation
BearbeitenDie Transkription der Schlüssel- oder Schrittmacherenzyme wird durch Glucose und Insulin gefördert und durch cAMP gehemmt.
Der erste und durch Insulin geförderte Schritt, die Umwandlung von Glucose in Glucose-6-phosphat durch das 1. Schlüsselenzym, die Hexokinase dient der Fixierung von Glucose in der Zelle, nachdem es aus dem Blut aufgenommen wurde. Glucose-6-phosphat kann die Zelle nicht mehr verlassen. Dadurch, dass Glucose aus dem Diffusionsgleichgewicht entfernt wird, kann vermehrt neue Glucose nachströmen. Unterstützt wird dieser Prozess durch die Translokation des Glucosetransporters GLUT4 in die Plasmamembran, die ebenfalls von Insulin gefördert wird. Glucose-6-phosphat steht dann intrazellulär für zahlreiche Stoffwechselwege zu Verfügung. Darunter die Glycolyse, der Hexosemonophosphatweg, der Inositolphosphat-Stoffwechsel und nach Umwandlung in UDP-Glucose können die C6-Körper in die Glycogensynthese, die Biosynthese der Uronsäuren und den Galactose-Stoffwechsel fließen.
Das 2. Schlüsselenzym, die 6-Phosphofruktokinase katalysiert die Bildung von Fructose-1,6-bisphosphat, welches im nächsten Schritt gespalten wird. Damit ist die Phosphofruktokinase für die Einleitung des endgültigen Glucoseabbaus verantwortlich und das wichtigste Stellglied der Glycolyse. Reguliert wird das Enzym durch den wichtigsten allosterischen Regulator, das D-Fructose-2,6-bisphosphat. Fructose-2,6-bisphosphat schaltet die Phosphofruktokinase und damit die Glycolyse an und gleichzeitig die Fructose-1,6-Bisphosphatase, das komplementäre Enzym der Gluconeogenese ab.
Das 3. Schlüsselenzym ist die Pyruvatkinase. Dieses Enzym katalysiert den letzten Schritt der Glycolyse und sorgt insbesondere für den Nettogewinn von 2 ATP pro Glucosemolekül. Gehemmt wird das Enzym allosterisch von Alanin und ATP. Diese Moleküle signalisieren dem Enzym ein ausreichendes Energieangebot.
Bedeutung der anaeroben Glycolyse
Bearbeiten| Cori- und Glucose-Alanin-Zyklus | ||||||||||||
| ||||||||||||
| Farbcode: Stickstoffaufnahme/-transport/-abgabe |
Der anaerobe Abbau von Glucose ist die einzige Möglichkeit, wie Zellen und Gewebe auch unter Sauerstoffmangel Energie erzeugen können, wenn auch vergleichsweise ineffizient (2 ATP gegenüber maximal 32 ATP bei vollständiger Oxidation der Glucose über Citratzyklus und Atmungskette). Das entstehende Lactat wird in die Umgebung resp. das Blut abgegeben. Netto kann der Gesamtorganismus anaerob keine Energie gewinnen, da das Lactat in der Leber wieder unter hohem Aufwand (6 ATP) zur Glucose aufgebaut werden muss. Anders als bei Bakterien, die Lactat in die Umgebung abgeben, akkumuliert das Produkt rasch im Körper und muss daher wegen seiner Azidogenität (pKs = 3,86) beseitigt werden, zweitens muss die Glucose regeneriert werden, da die Vorräte sonst rasch erschöpft sind.
Der anaerobe Glucoseabbau zur Energiegewinnung spielt für den Skelettmuskel unter erhöhter Belastung eine große Rolle, wenn mehr Lactat entsteht als oxidativ in den Mitochondrien weiterverarbeitet werden kann. Das entstehende Lactat wird über den Blutweg zur Leber transportiert, dort wieder zur Glucose aufgebaut und dann zum Muskel zurücktransportiert (Cori-Zyklus). Der Abbau bis zum Lactat hat den Vorteil, dass die NADH-Bilanz neutral bleibt und damit auch das Redoxgleichgewicht in der Muskel- und Leberzelle.
Die zweite Möglichkeit besteht darin, Pyruvat durch Stickstoffübertragung in Alanin umzuwandeln (Transaminierung durch GPT bzw. AL(A)T und Pyridoxalphosphat), dieses zur Leber zu transportieren, dort wieder zu Pyruvat zu deaminieren (das frei werdende Ammoniak kann in der Leber zu Harnstoff entgiftet und über die Niere ausgeschieden werden) und wieder Glucose zu bilden. Der letztgenannte Weg, der Glucose-Alanin-Zyklus, ist wichtig, da hiermit auch der Stickstoff (-> giftiges Ammoniak) aus dem Muskel zur Leber geschafft wird, der durch die gesteigerte Proteolyse (Desaminierung glucogener Aminosäuren) vermehrt freigesetzt wird.
Erythrozyten nutzen den anaeroben Abbau von Glucose, da sie keine Mitochondrien haben. Bei lokalem Sauerstoffmangel greifen alle betroffenen Zellen auf dieses „Notstromaggregat“ zurück, um zu überleben.
Verbindungen zu anderen Stoffwechselwegen
BearbeitenSiehe dazu die Stoffwechsel-Übersicht.
Glucose wird in die Zelle aufgenommen und (wenn es nicht für die Fructose-Bildung (v.a. in den Samenblasen) genutzt wird) in Glucose-6-phosphat umgewandelt, so dass es die Zelle nicht mehr verlassen kann. Glucose-6-phosphat kann dann in der Glycolyse zu Pyruvat abgebaut werden oder je nach Organ und Bedarf für die Bildung von Glycogen (Leber, Skelettmuskel), Galactose (u.a. Milchdrüse), Glucuronsäure (v.a. Leber) oder Inositolphosphate verwendet werden sowie in den Pentosephosphatweg fließen. Umgekehrt ist Glucose-6-phosphat das Endprodukt des Abbaus von Glycogen (Leber, Muskel) und Galactose.
Die nächste Station ist Fructose-6-phosphat. Hier mündet bereits wieder ein Teil des Substrats des Pentosephosphat-Shunts ein (bzw. fließt dorthin ab). F6P stellt weiterhin die Verbindung zum Mannose- und Fucose-Stoffwechsel sowie zum Aminozucker-Stoffwechsel her.
Die isomeren Spaltprodukte von Fructose-1,6-Bisphosphat - Glycerinaldehyd-3-phosphat und Dihydroxyacetonphosphat - liefern das Glycerin für die Biosynthese der Neutralfette und Phosphoglyceride. Umgekehrt kann das Glycerin aus der Lipolyse hier in die Glycolyse/Gluconeogenese einmünden. Der Abbau von Fructose liefert ebenfalls DHAP und Glycerin und das 2. Endprodukt (bzw. Edukt für die Gegenrichtung) des Pentosephosphatwegs ist Glycerinaldehyd-3-phosphat. Die Verbindungen sind hier nocheinmal dargestellt.
3-Phosphoglycerat ist der Startpunkt der Serin- und Glycin-Biosynthese.
Phosphoenolpyruvat (PEP) wird für die Synthese von N-Acetylneuraminsäure (NANA) benötigt.
Pyruvat kann durch Transaminierung in Alanin umgewandelt werden und durch Reduktion in Lactat sowie vice versa. Pyruvat ist weiterhin der Ausgangpunkt der Gluconeogenese und die mögliche Endstrecke des Abbaus von Cholin und einigen glucogenen Aminosäuren wie dem genannten Alanin, Tryptophan (über Alanin), Threonin, Serin, Glycin und Cystein (über Serin). Pyruvat kann weiterhin aus dem Citratzyklus bezogen werden (Decarboxylierung von Malat) und zu guter letzt zur Energiegewinnung oder zur Biosynthese von Fettsäuren oder Cholesterin durch dehydrierende Decarboxylierung zu Acetyl-CoA umgesetzt werden.
Transport der Reduktionsäquivalente ins Mitochondrium zur Atmungskette
BearbeitenNADH (+ H+) kann die die innere Mitochondrienmembran nicht durchdringen. Die in der Glycolyse erzeugten Reduktionsäquivalente müssen jedoch irgendwie vom Zytosol ins Mitochondrium zur Atmungskette gelangen. Zur Lösung dieses Problems gibt es verschiedene Möglichkeiten.
Das Malat-Aspartat-Shuttle
Bearbeiten| ⇓ | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| NAD+ | 2. |
NAD+ | Malat-Dehydrogenase | 1.1.1.37 | Ox | |||||
|
||||||||||
| L-Glutamat | 3. |
L-Glutamat | Pyridoxal- phosphat | Aspartat-Transaminase (AST, ASAT, GOT) |
2.6.1.1 | Tr | ||||
|
||||||||||

Eine geschickte Lösung bietet die Übertragung der Elektronen auf ein Transportmolekül, in diesem Falle auf Oxalacetat, das dadurch zum Malat reduziert wird (1.). Malat wird nun in die Matrix geschafft, wo die Malat-Dehydrogenase die entsprechende Rückreaktion katalysiert, so dass die Elektronen wieder auf NAD+ übertragen werden (2.).
Nun wird der Rückweg ins Zytosol dadurch verkompliziert, dass Oxalacetat die innere Mitochondrienmembran ebenfalls nicht permeieren kann. Deswegen muss Oxalacetat in eine Form gebracht werden, für die ein Membrantransporter vorhanden ist. Oxalacetat wird also von der Aspartat-Transaminase (AST, GOT) PALP-abhängig zu Aspartat transaminiert (3). Die Aminogruppe stammt dabei von Glutamat, das durch die Transaminierung zum α-Ketoglutarat wird. Aspartat und α-Ketoglutarat gelangen nun über die entsprechenden Membrantransporter zurück ins Zytosol, die Aspartat-Transaminase sorgt noch einmal für die Rückreaktion (4.) und es entsteht wieder Oxalacetat für den nächsten Transportzyklus, sowie Glutamat, das wieder in die Matrix transportiert wird und dort für die nächste Transaminierung bereitsteht.
Das Zusammenspiel aus Umwandlungs- und Transportprozessen ergibt eine pfiffige Choreographie, die in der Abbildung rechts noch einmal schematisch dargestellt ist.
Die beteiligten Reaktionspartner Oxalacetat, Malat und α-Ketoglutarat sind (unter anderem) Intermediate des Citratzyklus.
Das Glycerin-3-Phosphat-Shuttle
Bearbeiten| ⇓ | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| NADH/H+ | 1. |
FADH2 | 1) Glycerin-3-phosphat-Dehydrogenase | 1.1.1.8 | Ox | |||||
| 2) ? | ? | Ox | ||||||||
|
||||||||||
Eine Alternative stellt das Glycerin-3-phosphat-Shuttle dar. Das Glycolyse-Intermediat DHAP wird dabei zu Glycerin-3-phosphat reduziert (1.) und überträgt sie an der inneren Mitochondrienmembran auf FAD (2.). FADH2 gibt die Elektronen an Ubichinon (Q) weiter. Das Glycerin-3-phosphat-Shuttle ist etwas schneller als das Malat-Aspartat-Shuttle, hat jedoch durch die Umgehung der NADH-Dehydrogenase (Komplex I der Atmungskette) eine etwas geringere Energieausbeute. Man findet dieses Shuttle z.B. im Gehirn und im Skelettmuskel.
In einem Nebenweg der Glycolyse entsteht 2,3-Bisphosphoglycerat
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
|
5.4.2.4 | Iso | BPGM-Def. | |||||||
|
||||||||||
| H2O
Pi |
|
3.1.3.13 | Hyd | |||||||
|
||||||||||
1,3-Bisphosphoglycerat und 3-Phosphoglycerat sind Intermediate der Glycolyse, wobei das letzteres aus ersterem unter ATP-Gewinn und katalysiert von der Phosphoglycerat-Kinase gebildet wird (s.o.).
1,3-Bisphosphoglycerat kann jedoch auch von der Bisphosphoglycerat-Mutase zu 2,3-Bisphosphoglycerat (2,3-BPG) isomerisiert werden. Dies spielt insbesondere in Erythrozyten eine wichtige Rolle. Dort verschiebt 2,3-BPG die Sauerstoffbindungskurve des Hämoglobins durch Stabilisierung des Desoxyhämoglobins nach rechts und erleichtert so die Sauerstoffabgabe im Gewebe.
Durch die Bisphosphoglycerat-Phosphatase-Reaktion kann 2,3-BPG es zu 3-Phosphoglycerat dephosphoryliert und damit wieder der Glycolyse zugeführt werden. Die Reaktionen laufen auch unter der Bezeichnung Rapoport-Luebering-Zyklus.
Im Gegensatz zur Phosphoglycerat-Kinase-Reaktion kann hierbei kein ATP gewonnen werden.
Pathobiochemie
BearbeitenEnzymdefekte der Glycolyse (bzw. Gluconeogenese) äußern sich häufig in hämolytischen Anämien, Myopathien und Neurodegeneration. Dies hat damit zu tun, dass Erythrozyten (keine Mitochondrien!) und Nervenzellen ihren Energiebedarf fast ausschließlich über Glucose decken, ebenso wie der Skelettmuskel unter anaeroben Bedingungen.
Klinische Chemie und Laboratoriumsmedizin
BearbeitenDie Umschaltung des Skelettmuskels auf den anaeroben Glucoseabbau zur Energiegewinnung unter Belastung, die sog. „anaerobe Schwelle“, ist als Lactat-Anstieg im Blut messbar. Für den Sport wird allgemein aerobes Training empfohlen. Ein erhöhtes Lactat ist auch bei schweren Allgemeinerkrankungen und ischämischen Zuständen feststellbar, wenn die Sauerstoffversorgung des Organismus oder einzelner Gewebe insuffizient wird, z.B. im Schock oder bei einer Darmischämie. Die sog. metabolische Laktatazidose geht mit einer lebensbedrohlichen Übersäuerung des Körpers einher und kann verschiedenste Ursachen haben, z.B. eine Vergiftung.
Die Serum-Konzentration der Lactat-Dehydrogenase (LDH) steigt bei einem erhöhten Zellzerfall und kann daher als Parameter für z.B. Gewebsschädigungen oder Tumorerkrankungen (hoher Zellumsatz) dienen. Eine unsachgemäße Blutentnahme (Hämolyse) kann dergleichen vortäuschen. Neben der LDH können auch erhöhte Harnsäure- (Endprodukt des Purin-Stoffwechsels (DNA, RNA)) und Kalium-Spiegel (normalerweise vorwiegend intrazellulär) auf Zelluntergänge hindeuten, wenn sie nicht durch andere Störungen (Niereninsuffizienz, Gicht, Medikamente) bedingt sind.
Weblinks
Bearbeiten- KEGG: Glycolysis / Gluconeogenesis - Homo sapiens (human)
- RCSB PDB: Glycolytic Enzymes
- The chemical logic behind... Glycolysis von Prof. Doutor Pedro Silva.
- Glycolysis in animation (von John Kyrk)
- Glycolysis (von Donald Nicholson)
- The Biochemists' Songbook MP3 Files: In Praise of E. M. P. (Tune: "The British Grenadiers")
Gluconeogenese
BearbeitenAllgemeines
BearbeitenDie Gluconeogenese (Glucose-Neubildung) ist eine energieaufwendige Möglichkeit, die Glycolyse umzukehren. Sie wird neben dem Glycogen-Abbau vor allem von der Leber dazu genutzt, um den Blutzuckerspiegel konstant zu halten.
Teil 2: Bildung von Glucose aus Glycerinaldehyd-3-phosphat (GADP) und Dihydroxyacetonphosphat (DHAP)
Bearbeiten| Tr. | All. | ( ⇓ ) | Substrat | ⇑ | Co. | Enzym | EC | EG | Erkr. | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||||
|
+ cAMP |
Pi
H2O |
Glucose-6- Phosphatase | 3.1.3.9 | Hyd | GSD1a (von Gierke) | |||||||||
|
||||||||||||||
|
5.3.1.9 | Iso | GPI-Def. | |||||||||||
|
||||||||||||||
|
+ cAMP |
- AMP, F-2,6-BP | Pi
H2O |
Fructose-1,6- bisphosphatase | 3.1.3.11 | Hyd | FBP-Def. | ||||||||
|
||||||||||||||
| Zn |
|
4.1.2.13 | Ly | GSD12, Hered. Fructoseintoleranz | ||||||||||
|
|
5.3.1.1 | Iso | TPI1-Def. | ||||||||||
Teil 1: (Rück-)Gewinnung von Glycerinaldehyd-3-phosphat aus Pyruvat
Bearbeiten| Tr. | All. | ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||
| Pi + NAD+ | NAD+ + Pi |
|
1.2.1.12 | Ox | ||||||||
|
||||||||||||
| ADP
ATP |
ADP
ATP |
|
2.7.2.3 | Tr | PGK1-Def. | |||||||
|
||||||||||||
|
5.4.2.1 | Iso | GSD10 | |||||||||
|
||||||||||||
|
|
|
Mg |
|
4.2.1.11 | Ly | ENO1-Def., GSD13 | ||||||
|
||||||||||||
|
+ cAMP, Gluko- kortikoide |
GDP, CO2
GTP |
Phosphoenolpyruvat- Carboxykinase | 4.1.1.32 | Ly | PCK1-Def., PCK2-Def. | |||||||
|
||||||||||||
| + Acetyl- CoA | ADP, Pi
ATP, HCO3- |
Biotin; Mn od. Zn | Pyruvat-Carboxylase | 6.4.1.1 | Lig | PC-Def. | ||||||
|
||||||||||||
| NADH/H+ | NADH/H+ | L-Lactat-Dehydrogenase | 1.1.1.27 | Ox | GSD11, LDHB-Def. | |||||||
|
||||||||||||

Die Gluconeogenese
BearbeitenDie Gluconeogenese stellt quasi die Umkehrung der Glycolyse dar. Aus zwei Pyruvat (Lactat) wird hier ein Glucosemolekül gebildet. Um den Substratfluss in die entgegengesetzte Richtung zu leiten, müssen dabei drei irreversible exergone Reaktionen der Glycolyse gegen endergone ausgetauscht werden. Nicht zufällig stellen diese Reaktionen bzw. die katalysierenden Enzyme auch die Schlüsselenzyme dar, über die zwischen Glycolyse und Gluconeogenese hin- und hergeschaltet wird. Da die Gluconeogenese sehr energieaufwendig ist – sie kostet 6 Mol ATP (und 2 NADH/H+) pro Mol Glucose, während die Glycolyse nur 2 ATP (und 2 NADH/H+) pro Mol Glucose liefert – wird sie streng nach Bedarf aktiviert.
Die beteiligten Enzyme sind bis auf die Pyruvatcarboxylase (anaplerotische Reaktion des Citratzyklus im Mitochondrium) und die Glucose-6-Phosphatase (glattes endoplasmatisches Retikulum) im Zytosol lokalisiert. D.h. die Glucosebildung verteilt sich auf drei zelluläre Reaktionsräume.
Die Gluconeogenese findet v.a. in Leber und Nierenrinde, z.T. auch in der Darmmucosa statt. Sie dient neben der Glycogenolyse dazu, den Blutzuckerspiegel anzuheben und glucoseabhängige Organe wie Nervensystem, Erythrozyten, Nebennierenmark und den arbeitenden Skelettmuskel mit Glucose zu versorgen. Angekurbelt wird die Gluconeogenese besonders unter Belastung bzw. Stress durch sympathische Katecholaminfreisetzung (cAMP-Anstieg im Hepatozyt) und Glucokortikoide sowie Glucagon. Dies erfolgt über die Beeinflussung der Transkriptionsrate sowie über die Senkung der Konzentration an Fructose-2,6-bisphosphat, dem wichtigsten allosterischen Regulator. Insulin ist der Gegenspieler und bremst die Gluconeogenese.
Substrate der Gluconeogenese sind Lactat (Cori-Zyklus), glucogene Aminosäuren, die bes. aus dem Skelettmuskel zufließen (Glucose-Alanin-Zyklus), und Glycerin, welches beim Abbau von Triglyceriden, also bei der Lipolyse entsteht, nach Aktivierung und Oxidation zu Dihydroxyacetonphosphat. Die Gluconeogenese aus Acetyl-CoA ist hingegen nicht möglich, daher können Fettsäuren, Ketonkörper und rein ketogene Aminosäuren auch nicht zur Gluconeogenese herangezogen werden!
Verbindungen zu anderen Stoffwechselwegen
BearbeitenDie Glycolyse der Nahrungsglucose bzw. Gluconeogenese aus Pyruvat, Lactat oder glucogenen Aminosäuren stellt als Rückgrat des Stoffwechsels zahlreichen anderen Stoffwechselwegen Substrat zur Verfügung und nimmt diese umgekehrt auch wieder auf. Die Verbindungen sind im Kapitel Glycolyse dargestellt. Der 1. Schritt der Gluconeogenese im Mitochondrium liefert anders als die Glycolyse zusätzlich noch Oxalacetat (Umgehung der Pyruvatkinase-Reaktion), womit der Citratzyklus (Mitochondrium) aufgefüllt werden kann bzw. umgekehrt kann das Oxalacetat aus dem Citratzyklus hier zur Gluconeogenese eingeschleust werden. Der Transport von Oxalacetat über die innere Mitochondrienmembran, die keinen Oxalacetat-Transporter hat, erfolgt z.B. in Form von Citrat (Kondensation von Oxalacetat mit Acetyl-CoA, 1. Reaktion des Citratzyklus) oder Malat (Reduktion von Oxalacetat zu Malat) mittels einem Tricarboxylatcarrier.
Pathobiochemie
BearbeitenBei der Glykogenspeicherkrankheit 1a (GSD1a, von Gierke) verhindert ein Defekt der Glucose-6-Phosphatase, dass die Leber Glucose aus der Gluconeogenese oder Glycogenolyse freisetzen kann, die Glucose bleibt als Glucose-6-phosphat in der Zelle gefangen. Schwere Hypoglykämien sind die Folge. Schwere Unterzuckerungen resultieren auch aus Defekten der Fructose-1,6-bisphosphatase und der Phosphoenolpyruvat-Carboxykinase 1 und 2, ebenfalls Schlüsselenzyme der Gluconeogenese. Die Defizienz des vierten Schrittmacherenzyms, der Pyruvat-Carboxylase, führt zu einer mangelnden Produktion von Oxalacetat. Aufgrund der vielfältigen Aufgaben von Oxalacetat kommt es hier zu komplexeren Störungsbildern.
Weblinks
Bearbeiten- KEGG: Glycolysis / Gluconeogenesis - Homo sapiens (human)
- The chemical logic behind... Gluconeogenesis von Prof. Doutor Pedro Silva
Glycogenolyse und Stärkeabbau
BearbeitenAllgemeines
BearbeitenDie Stärke der Pflanzen (Amylose und Amylopectin) und das tierische Glycogen stellen die kompakte Speicherform der Glucose dar. Sie bestehen aus langen Ketten von 1,4-α-verbundenen Glucosemolekülen, die sich über zusätzliche 1,6-α-gebundene Glucose-Moleküle verzweigen.
In Leber und Muskel gebildetes Glycogen dient im Körper als schnell verfügbarer Energie- bzw. Glucosespeicher.
Glycogen und Stärke aus der Nahrung werden im Verdauungstrakt von Amylasen, Glucosidasen und Isomaltasen zerlegt und dienen als exogene Kohlenhydratquelle.
Abbau von Kettenverzweigungen
Bearbeiten| ⇓ | Substrat | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
|
α-Glucantransferase |
2.4.1.25 | Tr | GSD3 (Forbe, Cori) | |||||||
|
||||||||||
| H2O
α-D-Glucose |
Amylo-1,6-Glucosidase |
3.2.1.33 | Hyd | GSD3 (Forbe, Cori) | ||||||
|
||||||||||



Zuerst wird die Amylose-Kette durch die α-Glucantransferase vom 1,6-gebundenen Glucose-Molekül (der Verzweigungsstelle) auf eine andere (bereits gekürzte) Amylose-Kette übertragen, also von einer 1,4- zu einer 1,4-Bindung. Das nun freistehende 1,6-gebundene Glucose-Molekül kann dann im 2. Schritt von der Amylo-1,6-Glucosidase abgespalten werden.
Freisetzung von Glucosemolekülen aus der (unverzweigten) Glycogenkette
Bearbeiten| Tr. | Kov. | All. | ⇓ | Substrat | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||
| + Phos- phory- lierung (Leber) |
+ AMP |
Pi Glc-[1,4-α-D- Glycosyl]n-1-Glc |
Pyrid- oxal- phos- phat |
|
2.4.1.1 | Tr | GSD5 (McArdle), GSD6 (Hers) | |||||
| Phospho- glucomutase | 5.4.2.2 | Iso | GSD14, CDG1T | |||||||||
|
+ cAMP |
H2O
Pi |
Glucose-6-Phosphatase | 3.1.3.9 | Hyd | GSD1a (von Gierke) | |||||||
|
||||||||||||
-alpha-D-Glucose.svg)

Die Freisetzung von Glucose-1-phosphat statt Glucose hat den Vorteil, dass die Glucose nicht erst wieder zum Glucose-6-phosphat mit ATP phosphoryliert werden muss, damit sie weiter verstoffwechselt werden kann. So kann ATP eingespart werden.
Glucoseabspaltung vom nicht-reduzierenden Kettenende her
BearbeitenSukkzessive Abspaltung terminaler 1,4-gebundener α-D-Glucose-Reste vom nicht-reduzierenden Ende her.
| ⇓ | Substrat | ⇑ | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| H2O
Glycogen/Dextrin |
Glucan-1,4-α-Glucosidase | 3.2.1.3 | Hyd | |||||||
|
||||||||||
| Aldose-1-Epimerase | 5.1.3.3 | Iso | ||||||||
|
||||||||||
| Oligosaccharid
H2O |
Lysosomale α-Glucosidase (saure Maltase) |
3.2.1.20 | Hyd | (GSD2) Pompe) | ||||||
| Intestinale Maltase-Glucoamylase | ||||||||||
|
||||||||||

1,4-α-Spaltung von Stärke und Glycogen zu Dextrin durch Amylase im Verdauungstrakt
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| H2O
[1,4-α-D-Glycosyl]n |
|
3.2.1.1 | Hyd | |||||||
|
||||||||||
1,6-α-Spaltung von 1,6-α-Bindungen im Verdauungstrakt
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| H2O
α-D-Glucose (oder Dextrin) |
Isomaltase | 3.2.1.10 | Hyd | |||||||
|
||||||||||

Eigenschaften von Glycogen und Stärke
BearbeitenPflanzliche Stärke setzt sich aus Amylose und Amylopectin zusammen. Amylose besteht aus Ketten von 250-300 Glucosemolekülen, die (wie Maltose auch) 1,4-α-glycosidisch verbunden sind. Amylopectin enthält zusätzlich noch an etwa jeder 25. Glucose eine 1,6-α-gebundene Glucose, wo sich dann die Kette verzweigt. Tierisches Glycogen entspricht weitgehend dem Amylopectin, ist allerdings noch stärker verzweigt (alle 6-10 Glucosereste). Durch die Verzweigungen entstehen große Makromoleküle.
Im tierischen Organismus findet der Auf- und Abbau von Glycogen vorwiegend in der Leber und im Muskel statt.
Abbau der Verzweigungen beim Glycogenabbau in Leber und Muskel
BearbeitenDie Entfernung der Verzweigungen (Tab. 1) erfolgt, wenn durch die benachbarte Glycogenolyse die Verzweigungsstelle für die debranching-Enzyme zugänglich geworden ist. Dann kann die α-Glucantransferase angreifen und die 1,6-gebundene Kette bis auf das 1,6-gebundene Glucosemolekül 1,4-glycosidisch auf die andere Kette übertragen. Das einzelne 1,6-gebundene Glucosemolekül kann nach der Exponierung der 1,6-Bindung nun durch die Amylo-1,6-Glucosidase abgespalten werden.
____ _
___/_ -> ___/_____ -> ________
α-Glucantransferase Amylo-1,6-Glucosidase
Abbau der 1,6-Verzweigungen im Verdauungstrakt
Bearbeitenα-1,6-glykosidische Bindungen werden im Dünndarm von der Isomaltase hydrolytisch gespalten (Tab. 5).
Abbau der Homopolysaccharidketten - 3 Varianten
Bearbeiten _ G1P _ Glc Bruchstücke
_____ ______ ____________ ____ _ _____ ___ _
____________ _____________ ______________
Phosphorylase Glucosidase α-Amylase
Aus der unverzweigten Glucose-Kette werden in Leber und Muskel sukzessive einzelne Glucosemoleküle direkt als Glucose-1-phosphat (ATP-Ersparnis!) von der Glycogen-Phosphorylase herausgeschnitten (Tab. 2) oder sie können durch die Glucosidase vom nicht-reduzierenden Ende her als Glucosemoleküle abgespalten werden (Tab. 3).
Mit der Nahrung aufgenommene pflanzlichen Stärke und Glycogen werden im Mund und Dünndarm durch α-Amylase (Tab. 4) und im Dünndarm durch α-Glucosidasen (Maltase, Tab. 3) an der α-1,4-glykosidischen Bindung hydrolytisch gespalten. Die α-Amylase hat ihr Aktivitätsmaximum im alkalischen Milieu und ist besonders im Sekret von Speicheldrüsen und Pankreas enthalten. Verbleibende α-1,6-glykosidische Bindungen werden von der Isomaltase abgebaut (Tab. 5).
Glycogenhaushalt
BearbeitenGlycogen wird bezogen auf das Gewicht am stärksten in der Leber gespeichert. Glycogen stellt eine leicht verfügbare Glucosereserve dar. Die Leber ist für die Kohlenhydrate und Aminosäuren aus dem Verdauungstrakt, die über die Pfortader antransportiert werden, das erste Auffangbecken. Da wir unregelmäßig essen, Organe wie das Gehirn aber ständig Glucose brauchen wird der Blutglucosespiegel streng kontrolliert. Die Leber ist hier das zentrale Regulationsorgan, in dem sie Glucose als Glycogen speichert und sie bei Bedarf wieder freisetzt und auch über die Gluconeogenese neu bildet. Bezogen auf den Körpergesamtglycogengehalt findet sich das meiste Glycogen allerdings in der Skelettmuskelmasse. Der Skelettmuskel besitzt keine Glucose-6-Phosphatase-Aktivität und nutzt seine Glycogenspeicher daher nur für den Eigenbedarf (Glucose-6-phosphat kann die Zelle nicht verlassen).
Regulation
BearbeitenDas Schlüsselenzym der Glycogenolyse ist die Glycogen-Phosphorylase. Allosterisch aktiviert wird das Enzym durch AMP, welches Energiemangel signalisiert. Allosterische Hemmer sind entsprechend ATP und Glucose-6-phosphat.
Die Leber-Isoform des Enzyms kann auch durch Phosporylierung eines Serin-Restes aktiviert werden. Die Phosphorylierung erfolgt durch die Phosphorylase-Kinase, die ihrerseits durch Phosphorylierung oder Ca2+-Calmodulin aktiviert wird. Aktivierung heißt es kommt zu einer Konformationsänderung des Enzyms. Die Glycogenolyse in der Leber wird stimuliert durch Glucagon (aus den A-Zellen des Pankreas) und Katecholamine (Adrenalin aus dem Nebennierenmark), die beide über membranständige G-Protein-gekoppelte Rezeptoren die Adenylatcyclase aktivieren (-> cAMP-Anstieg), sowie durch Glucokortikoide (aus der Nebennierenrinde). Insulin (aus den B-Zellen des Pankreas) wirkt als Gegenspieler und bremst den Glycogenabbau.
Klinische Chemie und Laboratoriumsmedizin
BearbeitenEine Erhöhung der α-Amylase im Serum kann auf eine Pankreatitis oder Sialadenitis hinweisen.
Pharmakologie
Bearbeiten
Oral zugeführte α-Glucosidase-Hemmer wie die Acarbose hemmen im Dünndarm die Spaltung von Stärke in Glucose. Beim Diabetes mellitus Typ 2 können damit postprandiale Blutzuckerspitzen verhindert werden. Gastrointestinale Nebenwirkungen kommen durch die bakterielle Zersetzung der nicht resorbierten Kohlenhydrate im Dickdarm zustande.
Weblinks
Bearbeiten- KEGG: Starch and sucrose metabolism - Homo sapiens (human)
- The chemical logic behind... Glycogen synthesis and degradation von Prof. Doutor Pedro Silva
- RCSB PDB: Glycogen Phosphorylase
- RCSB PDB: Alpha-amylase
Glycogensynthese
BearbeitenAllgemeines
BearbeitenGlykogen bildet v.a. in Leber und Skelettmuskel ein leicht verfügbares Glucose-Depot.
Verlängerung der Glycogenkette um ein Glucosemolekül sowie Einbau von Kettenverzweigungen
Bearbeiten| Kov. | ( ⇓ ) | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||
| Amylo-(1,4->1,6)- Transglucosylase (branching enzyme) | 2.4.1.18 | Tr | GSD IV (Andersen) | ||||||||
|
|||||||||||
| - Phos- phory- lierung | UDP
[1,4-α-D-Glycosyl]n |
Glycogen-Synthase |
2.4.1.11 | Tr | GSD 0a, GSD 0b | ||||||
| PPi | UTP-Glucose- 1-phosphat- Uridyltransferase | 2.7.7.9 | Tr | ||||||||
| Phospho-glucomutase | 5.4.2.2 | Iso | GSD14, CDG1T | ||||||||

Erzeugung neuer Glycogenketten
BearbeitenBei der Glycogensynthese werden entweder bestehende Ketten verlängert (s.o.) oder die Glycogensynthese beginnt mit dem Protein Glycogenin als Startermolekül, das sich selbst bis max. 13 Glucosereste anhängen kann:
| ( ⇓ ) | Substrat | ⇑ | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
|
|||||||
| UDP
UDP-Glucose |
Glycogenin-Glycosyltransferase |
2.4.1.186 | Tr | ||||
|
|||||||
| UDP
Glycogenin |
Glycogenin-Glycosyltransferase |
2.4.1.186 | Tr | ||||
|
|||||||
glycogenin.svg)

Weblinks
Bearbeiten- KEGG: Starch and sucrose metabolism - Homo sapiens (human)
- The chemical logic behind... Glycogen synthesis and degradation von Prof. Doutor Pedro Silva
Hexosemonophosphatweg
BearbeitenAllgemeines
BearbeitenDer Hexosemonophosphatweg (HMP-Weg, Pentosephosphatweg, Pentosephosphat-Shunt) ist ein Nebenweg der Glycolyse. Er dient der Reduktion von NADPH + H+ für Reduktionsvorgänge und der Biosynthese von Ribose-5-phosphat für die Nukleotid- resp. Purin- und Pyrimidin-Biosynthese einschl. Purin-Salvage.
1. Teil: Oxidation und Decarboxylierung von α-D-Glucose-6-phosphat zu Ribulose-5-phosphat
Bearbeiten| All. | ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | ||
|---|---|---|---|---|---|---|---|---|---|---|
| 6-Phospho- gluconat |
|
5.3.1.9 | Iso | Glucose-6-phosphat-Isomerase-Defizienz | ||||||
|
||||||||||
| NADP+ | Glucose-6-phosphat- 1-Dehydrogenase (G6PD) | 1.1.1.49 | Ox | Glucose-6-phosphat-Dehydrogenase-Mangel | ||||||
|
||||||||||
| H2O
|
6-Phosphogluconolactonase | 3.1.1.31 | Hyd | |||||||
|
||||||||||
| NADP+ | 6-Phosphogluconat- Dehydrogenase | 1.1.1.44 | Ox | 6-Phosphogluconat-Dehydrogenase-Defizienz | ||||||
|
||||||||||
|
|
||||||||||
|
||||||||||





2. Teil: Aus Ribulose-5-phosphat wird Fructose-6-phosphat und Glycerinaldehyd-3-phosphat
Bearbeiten| Substrat 1 | ⇓ ⇑ | Substrat 2 | Co. | Enzym(e) | EC | EG | Erkr. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
||||||||||||
| 1. |
2. |
1) Ribulose- phosphat-3-Epimerase
2) Ribose-5- phosphat-Isomerase |
1) 5.1.3.1
2) 5.3.1.6 |
Iso | 2) Ribose-5-phosphat-Isomerase-Defizienz | ||||||||
|
+ |
|
|||||||||||
| Thiamin- P2 | Transketolase | 2.2.1.1 | Tr | Wernicke-Korsakoff-Syndrom | |||||||||
|
+ |
|
|||||||||||
| Transaldolase | 2.2.1.2 | Tr | Transaldolase-Defizienz | ||||||||||
|
+ |
|
|||||||||||
| ---- | |||||||||||||
|
+ | ||||||||||||
| Thiamin- P2 | Transketolase | 2.2.1.1 | Tr | Wernicke-Korsakoff-Syndrom | |||||||||
|
+ |
|






Die Reaktionen im Detail
BearbeitenDer Hexosemonophosphatweg (HMP-Weg, Pentosephosphatweg) ist ein Nebenweg der Glycolyse und wie diese im Zytosol lokalisiert.
Phase 1 (Oxidative, nicht-reversible Phase):
- Zuerst wird α-D-Glucose-6-phosphat zu β-D-Glucose-6-phosphat isomerisiert.
- Die Glucose-6-phosphat-1-Dehydrogenase katalysiert danach die Oxidation am C1-Atom. Dabei wird ein NADPH/H+ gewonnen und es entsteht 6-Phosphogluconolacton.
- Durch Wasseraufnahme wird daraus unter Ringöffnung 6-Phosphogluconat.
- 6-Phosphogluconat kann nun ein weiteres Mal unter NADPH/H+-Gewinn oxidiert werden. Dabei entsteht ein instabiles Zwischenprodukt, das spontan zu Ribulose-5-phosphat decarboxyliert.
In der Summe wird also eine (Phospho-)Hexose zweimal oxidiert und zur (Phospho-)Pentose decarboxyliert.
Phase 2 (Nicht-oxidative, reversible Phase):
- Ribulose-5-phosphat wird sowohl zu Xylulose-5-phosphat als auch zu Ribose-5-phosphat isomerisiert.
- Beide Produkte können von der Transketolase zu D-Glycerinaldehyd-3-phosphat und Sedoheptulose-7-phosphat umgesetzt werden.
- Diese wiederum können mit Hilfe der Transaldolase zu β-D-Fructose-6-phosphat und Erythrose-4-phosphat reagieren.
- In einem weiteren Schritt kann Erythrose-4-phosphat unter Einfluss der Transketolase noch einmal mit Xylulose-5-phosphat (s.o.) in D-Glycerinaldehyd-3-phosphat und β-D-Fructose-6-phosphat umgewandelt werden, die beiden Endprodukte dieses Weges.
Im Endeffekt werden im HMP-Weg jeweils drei Glucosephosphat-Moleküle (3 C6-Körper = 18 C-Atome) decarboxyliert (es bleiben 18 - 3 = 15 C-Atome) und diese dann zu zwei Fructose-6-phosphat (2 C6-Körper = 12 C-Atome) und einem Glycerinaldehyd-3-phosphat (1 C3-Körper) umgesetzt.
Glycerinaldehyd-3-phosphat und Fructose-6-phosphat können wieder in die Glycolyse eingeschleust werden oder über die Gluconeogenese zu α-D-Glucose-6-phosphat umgesetzt werden. Durch letzteres entsteht ein Kreislauf, über den Glucose netto vollständig decarboxyliert werden kann. Pro Glucose-Molekül werden dabei 12 NADPH/H+ gewonnen.
Bedeutung des Hexosemonophosphatweges
Bearbeiten- Generierung von NADPH/H+ im 1. Teil:
- für NADPH-abhängige Biosynthesen wie z.B. Fettsäuren und Cholesterin.
- Insbesondere in den Erythrozyten wird das NADPH benötigt, um die Glutathionreduktase zu regenerieren. Dieses Enzym reduziert Glutathion, ein Tripeptid aus Glutamat, Glycin und Cystein, welches mit seiner reduzierten Sulfhydrylgruppe (SH-Gruppe von Cystein) Hämoglobin u. a. Proteine vor der Oxidation schützt.
- Generierung von Pentosen wie D-Ribose-5-phosphat z. B. für die Nucleotid- und Nucleinsäuresynthese. Dafür kann der 2. Teil des Hexosephosphatweges von D-Glycerinaldehyd-3-phosphat und D-Fructose-6-phosphat ausgehend auch einfach rückwärts ablaufen, wenn z.B. Pentosen, aber kein NADPH/H+ benötigt wird.
Regulation
BearbeitenDer 1. Teil des HMP-Weges wird reguliert durch das Angebot an NADP+ (Aktivierung) und NADPH/H+ (Hemmung). Das Schrittmacherenzym ist dabei die Glucose-6-phosphat-Dehydrogenase. Die Reaktionen des 2. Teils sind reversibel und laufen entsprechend dem Angebot an Substraten von unten (aus der Glycolyse/Gluconeogenese) oder oben (aus dem oxidativen Teil) ab und in dem Maße, wie Ribose-5-phosphat aus dem Gleichgewicht entfernt wird bzw. in die Nucleotid-Biosynthese abfließt.
Pathobiochemie
BearbeitenDer G6PD-Mangel führt bei oxidativem Stress mit H2O2-Bildung (Infektionen, Medikamente wie ASS, Sulfonamide, Malariamittel, Lebensmittel wie Saubohnen/Favabohnen) zur oxidativen Schädigung des Erythrozyten und zu hämolytischen Krisen (Favismus).
Weblinks
Bearbeiten- KEGG: Pentose phosphate pathway - Homo sapiens (human)
- The chemical logic behind... the Pentose-phosphate Pathway. By Prof. Doutor Pedro Silva.
Uronsäuren-Stoffwechsel
BearbeitenAllgemeines
BearbeitenGlucuronsäure wird an Eigen- und Fremdmoleküle geheftet, um ihre Wasserlöslichkeit und damit ihre Ausscheidungsfähigkeit zu erhöhen. Daneben kann sie zum Monosaccharid Xylose decarboxyliert werden. Beide gehen in die Biosynthese der Glycosaminoglycane (Proteoglycane) ein.
Synthese und Übertragung von Glucuronat
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
| Phosphoglucomutase | 5.4.2.2 | Iso | GSD14, CDG1T | |||||||
| UTP
PPi |
UTP-Glucose-1-phosphat- Uridylyltransferase | 2.7.7.9 | Tr | |||||||
| H2O, 2 NAD+
2 NADH/H+ |
2 NAD+, H2O
2 NADH/H+ |
UDP-Glucose-6- Dehydrogenase | 1.1.1.22 | Ox | ||||||
|
||||||||||
| Akzeptor
UDP |
Akzeptor
UDP |
Glucuronosyltransferase Glattes ER | 2.4.1.17 | Tr | Hyperbilirubinämie I (Gilbert-S.), Crigler- Najjar-S. Typ I und II | |||||
|
||||||||||
| H2O
Akzeptor-OH |
β-Glucuronidase | 3.2.1.31 | Hyd | Mucopolysaccharidose VII | ||||||
|
||||||||||



Glucuronsäure entsteht formell aus Glucose durch Oxidation am C6-Atom. Um das Glucuronat später besser auf Akzeptormoleküle übertragen zu können muss es mit UDP aktiviert sein. Die Synthese von UDP-Glucose steht auch am Beginn der Glycogen- und Galactose-Biosynthese.
In der Reaktionsfolge wird zuerst ein Glucose-6-phosphat zum Glucose-1-phosphat isomerisiert. Das C1-Atom kann mit seiner Phosphatgruppe nun leicht mit dem Phosphat von UTP unter Abspaltung von Pyrophosphat reagieren, so dass UDP-Glucose entsteht. Zweitens wird durch die Isomerisierung das C6-Atom für die Oxidation zugänglich. Das UDP-Glucuronat kann den Glucuronatrest anschließend auf andere Moleküle übertragen.
Durch die Kopplung von Molekülen an Glucuronsäure kann deren Wasserlöslichkeit erhöht werden. Sie erfolgt z.B. an Hydroxyl- oder Amino-Gruppen. Die Glucuronidierung ist (neben der Acetylierung u.a.m.) ein Mechanismus der hepatischen Biotransformation Phase II, mit der Substanzen wie z.B. Bilirubin wasserlöslicher gemacht werden, die über Galle und Darm oder über die Niere ausgeschieden werden sollen.
Glucuronat findet sich weiterhin als Struktur-Bestandteil in den Proteoglykanen Chondroitinsulfat und Heparansulfat. Hier wird D-Glucuronat teilweise zu L-Iduronat epimerisiert.
Glucuronat wird auch beim Abbau von Inositol gebildet.
Aus UDP-Glucuronat kann UDP-D-Xylose gebildet werden
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
|
|
FAD | UDP-Glucuronat-Decarboxylase | 4.1.1.35 | Ly | ||||||
|
||||||||||

Xylose (Holzzucker) wird benötigt für die Biosynthese von Chondroitinsulfat und Heparansulfat. Dabei ist Xylose der erste Zucker, der an den Serin-Rest des Proteinanteils o-glycosidisch gebunden wird.
Xylose wird im Körper nicht abgebaut, sondern unverändert ausgeschieden.
Weblinks
Bearbeiten- KEGG: Pentose and glucuronate interconversions - Homo sapiens (human)
- KEGG: Starch and sucrose metabolism - Homo sapiens (human)
Galactose-Stoffwechsel
BearbeitenAllgemeines
BearbeitenLactose (Milchzucker) bzw. Galactose (Schleimzucker) ist für alle Säugetiere (Mammalia) eine wichtige Energiequelle in der Neugeborenen- und Säuglingsperiode. Galactose wird zudem für den Aufbau der Glycane benötigt, die sich z.B. im Bereich der Schleimhäute finden. Ein Lactasemangel ist eine häufige Ursache für die Unverträglichkeit von Milch- und Milchprodukten im Erwachsenenalter. Enzymdefekte des Galactoseabbaus führen zur Galactosämie.
Abbau von Lactose bzw. Galactose zu Glucose (Leloir pathway)
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| H2O
α-D-Glucose |
β-Galactosidase | 3.2.1.23 | Hyd | GM1-Gangliosidose, Mucopolysaccharidose IVb (Morquio B) | ||||||
| Lactase | 3.2.1.108 | Lactose-Intoleranz | ||||||||
|
||||||||||
| ATP
ADP |
Galactokinase | 2.7.1.6 | Tr | Galaktosämie II | ||||||
|
||||||||||
| UDP-Glucose | UDP-Glucose | Galactose-1-phosphat-Uridylyltransferase | 2.7.7.12 | Tr | Galactosämie | |||||
|
||||||||||
| NAD | UDP-Glucose-4-Epimerase | 5.1.3.2 | Iso | Galactosämie III | ||||||
|
||||||||||



Lactose (Milchzucker) ist ein reduzierender Doppelzucker bei dem die zwei Monosaccharide β-glycosidisch miteinander verbunden sind. Lactose wird im Darm von Lactase (β-Galactosidase) hydrolytisch in die Einzelzucker Glucose (Traubenzucker) und Galactose (Schleimzucker) gespalten.
Die Galactose kann dann in drei Schritten (Leloir pathway) zu UDP-Glucose umgewandelt werden:
- Galactose wird von der Galactokinase zu Galactose-1-phosphat phosphoryliert.
- Dieses kann wiederum von der Galactose-1-phosphat-Uridylyltransferase katalysiert ein UMP von UDP-Glucose übernehmen, wobei Glucose-1-phosphat frei wird.
- Die nun aktivierte UDP-Galactose kann dann von der UDP-Glucose-4-Epimerase zu UDP-Glucose epimerisiert werden (Galactose unterscheidet sich von Glucose letztlich nur in der Stellung der Hydroxylgruppe am C4-Atom).
Biosynthese von Lactose aus Glucose
Bearbeiten| ⇓ | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| NAD | UDP-Glucose-4-Epimerase | 5.1.3.2 | Iso | Galactosämie III | ||||||
|
||||||||||
| α-D-Glucose
UDP |
Lactose-Synthase | 2.4.1.22 | Tr | |||||||
|
||||||||||
Die Lactosebiosynthese in der lactierenden Brustdrüse wird von einem Enzymkomplex aus β-1,4-Galactosyltransferase (beta4Gal-T1) mit α-Lactalbumin katalysiert. Hierbei wird UDP-Galactose unter Abspaltung von UDP über eine β-1,4-glycosidische Bindung mit Glucose zur Lactose verbunden(s.o.).
UDP-Galactose wird zudem für die Biosynthese verschiedener Glycane, z.B. Chondroitinsulfat verwendet, das reichlich im Bereich der Schleimhäute zu finden ist, daher die deutsche Bezeichnung „Schleimzucker“ für Galactose.
Umwandlungen von Glucose, Glucose-phosphat und UDP-Glucose
Bearbeiten| Tr. | All. | ⇓ | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||
| - G6P | ATP
ADP |
1. |
Pi
H2O |
1) Hexokinase | 2.7.1.1 | Tr | Hexokinase I-Defizienz | |||||
| + Insulin | - GKRP | 1) Glucokinase (HK IV) | 2.7.1.2 | MODY2, PNDM, HHF3 | ||||||||
| + cAMP
- Insulin |
2) Glucose-6- Phosphatase | 3.1.3.9 | Hyd | GSD 1a (von Gierke) | ||||||||
| Phosphoglucomutase | 5.4.2.2 | Iso | GSD14, CDG1T | |||||||||
| UTP
PPi |
1. |
UMP
H2O |
1) UTP-Glucose-1-phosphat- Uridylyltransferase | 2.7.7.9 | Tr | |||||||
| 2) Nucleotid- Diphosphatase | 3.6.1.9 | Hyd | ||||||||||
|
||||||||||||
Hier sind noch einmal die Umwandlungsmöglichkeiten von der Glucose bis zur UDP-Glucose in beide Richtungen abgebildet. Die Hexokinase gehört zur Glycolyse, die Glucose-6-Phosphatase zur Gluconeogenese. Die Bildung des aktivierten Zuckers steht auch am Anfang der Biosynthese von Glycogen und Glucuronsäure.
Pathobiochemie
BearbeitenAbhängig von Ethnie und Lebensalter ist die Lactase-Aktivität im Darm mitunter nicht ausreichend um die Lactose von Milchprodukten vollständig zu spalten. Lactose wird nicht resorbiert und führt einerseits zum osmotischen Wassereinstrom, andererseits kann es zur bakteriellen Zersetzung im Dickdarm kommen. Typische Folgen des Lactase-Mangels (Milchzuckerunverträglichkeit) sind gastrointestinale Beschwerden wie Blähungen und Durchfall.
In Asien und Afrika sind mehr als 90 % der Erwachsenen betroffen, Kaukasier zu 5 bis 15 %. Mit dem Alter nimmt die Lactase-Aktivität natürlicherweise ab, da Milch in der Tierwelt nur der Säuglingsernährung dient.
Ein Lactase-Mangel kann allerdings auch primär vorhanden sein bedingt durch einen Gendefekt. Auch chronische Magen-Darm-Erkrankungen können die Lactoseverwertung beeinträchtigen.
Als Behandlungsansatz bietet sich je nach Restkapazität eine Lactosearme oder -freie Diät an. Auch orale Enzymersatzpräparate sind erhältlich.
Alle drei Enzyme des Leloir pathway können defizient sein und zum Krankheitsbild der Galactosämie führen. Eine Defizienz der Galactose-1-phosphat-Uridylyltransferase (GALT) ist die häufigste Ursache der Galactosämie („klassische Galactosämie“). Die Häufigkeit beträgt etwa 1:47.000 Geburten. Die Erkrankung wird mit einer streng Galactose-freien Diät behandelt, nichtsdestotrotz können Spätfolgen wie kognitive oder ovarielle Störungen auftreten. Ursache ist wohl die endogene Galactosebiosynthese aus anderen Zuckern.
Literatur
Bearbeiten- Fridovich-Keil JL. “Galactosemia: the good, the bad, and the unknown”. J. Cell. Physiol., 209:701–5, December 2006. DOI:10.1002/jcp.20820. PMID 17001680.
- Holden HM, Rayment I, Thoden JB. “Structure and function of enzymes of the Leloir pathway for galactose metabolism”. J. Biol. Chem., 278:43885–8, November 2003. DOI:10.1074/jbc.R300025200. PMID 12923184.
- Frey PA. “The Leloir pathway: a mechanistic imperative for three enzymes to change the stereochemical configuration of a single carbon in galactose”. FASEB J., 10:461–70, March 1996. PMID 8647345.
Weblinks
Bearbeiten- KEGG: Galactose metabolism - Homo sapiens (human)
- KEGG: Glycolysis / Gluconeogenesis - Homo sapiens (human)
Fructose-, Mannose- und Fucose-Stoffwechsel
BearbeitenSaccharose wird im Darm in Glucose und Fructose gespalten
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||||
| H2O
|
α-Glucosidase (saure Maltase) | 3.2.1.20 | Hyd | GSD II (Pompe) | |||||||||
| Sucrose-α-Glucosidase (Sucrase-Isomaltase) | 3.2.1.48 | Hyd | CSID | ||||||||||
|
|||||||||||||


Saccharose (Sucrose) ist als Haushalts- bzw. Kristallzucker reichlich in unserer Nahrung vorhanden. Im Darm erfolgt die Spaltung der α-1,2-glykosidischen Bindung des nicht-reduzierenden Disaccharids in Invertzucker, d.h. in äquimolare Mengen an Glucose (Traubenzucker) und Fructose (Fruchtzucker).
Biosynthese von Fructose aus Glucose (Polyolsyntheseweg)
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
| NADPH/H+ | NADPH/H+ | Aldehyd-Reduktase | 1.1.1.21 | Ox | |||
|
|||||||
| NAD(P)+ | NAD(P)+ | Sorbitol-Dehydrogenase | 1.1.1.14 | Ox | |||
|
|||||||

Fructose kann in extrahepatischen Geweben im Polyolsyntheseweg aus Glucose gebildet werden. Ein Zwischenprodukt ist der Polyalkohol (Zuckeralkohol) Sorbitol. Eine hohe Fructoseproduktion findet unter dem Einfluss von Testosteron in den Samenblasen statt.
In der Leber wird Fructose zu DHAP und D-Glycerinaldehyd abgebaut
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||||
| ATP
ADP |
Ketohexokinase | 2.7.1.3 | Tr | KHK-Def. (Fructosurie) | |||||||||
|
|||||||||||||
|
|
Zn | Fructose-1,6-bisphosphat-Aldolase | 4.1.2.13 | Ly | Fructoseintoleranz (Aldolase B-Def.), Aldolase A-Def. (Hämolyt. Anämie, Myopathie, Dysmorphie) | |||||||
|
|||||||||||||
| NAD(P)H/H+ | NAD(P)H/H+ | Zn / Fe | Alkohol-Dehydrogenase (NAD+) | 1.1.1.1 | Ox | ||||||||
| Zn | Alkohol-Dehydrogenase (NADP+) | 1.1.1.2 | Ox | ||||||||||
| Aldehyd-Reduktase (NAD(P)+) | 1.1.1.21 | Ox | |||||||||||



Die Fructose aus der Nahrung wird überwiegend in der Leber abgebaut.
Dihydroxyacetonphosphat kann in die Glycolyse bzw. Gluconeogenese eingehen, sowie in die Triacylglycerin-Biosynthese und revers in den Hexosemonophosphatweg. D-Glycerinaldehyd steht nach Oxidation zum Glycerin für die Triacylglycerin-Biosythese zur Verfügung oder kann ebenfalls in Dihydroxyacetonphosphat (DHAP) umgewandelt werden.
Der extrahepatische Abbau erfolgt nach Phosphorylierung von Fructose zu Fructose-6-phosphat
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
|
|||||||
| ATP
ADP |
Hexokinase | 2.7.1.1 | Tr | Hämolyt. Anämie bei HK1-Def. | |||
Fructose-6-phosphat ist ebenfalls wieder ein Intermediat der Glycolyse. Dieser Abbauweg findet z.B. im Muskel statt. In der Leber ist er nicht möglich, da dort die Hexokinase IV (Glucokinase) dominiert, die nur eine geringe Affinität zur Fructose hat.
Pathobiochemie des Fructose-Stoffwechsels
BearbeitenDie häufigste Ursache einer Fruktosunverträglichkeit ist die Fruktosemalabsorption, die wahrscheinlich auf eine Insuffizienz eines intestinalen Zuckertransporters zurückgeht (GLUT-5 ?). Die Fruktose wird im Darm bakteriell vergoren und führt zu Bauchschmerzen, Blähungen und Diarrhoe. Die Unverträglichkeit kann mit einem Atemtest diagnostiziert werden. Die Behandlung besteht in einer entsprechenden Diät.
Die Fructosurie ist eine gutartige, i.d.R. asymptomatische „Erkrankung“. Ihr liegt ein Defekt der Fructokinase resp. Ketohexokinase zugrunde.
Die seltene autosomal-rezessive hereditäre Fruktoseintoleranz ist zurückzuführen auf einen Defekt der Aldolase B, die v.a. in Leber, Niere und Dünndarmmucosa exprimiert wird. Infolge der fehlenden enzymatischen Aktivität kommt es bei Fruktose- oder Sorbitol-Exposition zur Akkumulation von Fructose-1-phosphat und Depletion an anorganischem Phosphat. Letzteres stimuliert die AMP-Deaminase, die AMP zu IMP deaminiert. IMP wird vermehrt abgebaut, was zwar Phosphat freisetzt, aber natürlich auch die Harnsäure-Bildung (Hyperurikämie) stimuliert und die AMP-Spiegel sinken lässt. Letzteres beeinträchtigt die oxidative Phosphorylierung (Regeneration von ATP). Ein weiteres Symptom sind Hypoglykämien, einerseits weil die Aldolase B in der Leber auch einen Schritt der Gluconeogenese katalysiert, andererseits weil die Glycogen-Phosphorylase und damit die Glycogenolyse wegen des AMP- und Phosphat-Defizits nur unzureichend stimuliert wird. Weitere Symptome sind Wachstumsretardierung und bes. nach fortgesetzter Fructoseexposition Azidose, Erbrechen, Leberschäden und Gelbsucht, Krämpfe und proximale tubuläre Azidose. Durch eine Fructose-freie Diät kann Symptomfreiheit erreicht werden.
Symptome der Aldolase A-Defizienz sind z.B. hämolytische Anämie, Myopathie und Dysmorphie, die u.a. durch eine Beeinträchtigung von Glycolyse und Gluconeogenese erklärt werden können.
Fructose, Mannose und Fucose
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| Zn | Mannose-6-phosphat- Isomerase | 5.3.1.8 | Iso | CDG1b | ||||||
|
||||||||||
| Phospho- mannomutase | 5.4.2.8 | Iso | CDG1a | |||||||
|
||||||||||
| GDP
Pi |
Mannose-1-phosphat- Guanylyltransferase | 2.7.7.22 | Tr | |||||||
|
||||||||||
|
|
NAD | GDP-Mannose-4,6- Dehydratase | 4.2.1.47 | Ly | ||||||
|
||||||||||
| NADPH/H+ | GDP-L-Fucose-Synthase | 1.1.1.271 | Ox | |||||||
|
||||||||||
| PPi
GTP |
PPi
GTP |
Fucose-1-phosphat- Guanylyltransferase | 2.7.7.30 | Tr | ||||||
|
||||||||||
| ADP
ATP |
dival. Kation | Fucokinase | 2.7.1.52 | Tr | ||||||
|
||||||||||






Die aktivierte Form der Mannose, die GDP-D-Mannose wird aus β-D-Fructose-6-phosphat gebildet, einem Intermediat der Glycolyse. Freie D-Mannose kann wie Fructose und Glucose auch von der Hexokinase (2.7.1.1) zu D-Mannose-6-phosphat phosphoryliert werden.
Aus GDP-D-Mannose wird die aktivierte Fucose, die GDP-L-Fucose, gebildet. Freie Fucose kann durch die Fucokinase und Fucose-1-phosphat-Guanylyltransferase reaktiviert werden.
Fucose und Mannose findet man als Strukturbestandteile häufig im Glycan-Anteil von Glycoproteinen.
Mannose-6-phosphat wird als „Adressierungs-Etikett“ im Golgi-Apparat an lysosomale Enzyme (Glykoproteine) angehängt (Proteintargeting).
Weblinks
Bearbeiten
Aminozucker-Stoffwechsel
BearbeitenAllgemeines
BearbeitenAminozucker finden sich v.a. in Glykanen. Ihr Syntheseweg beginnt mit Fructose-6-phosphat, einem Intermediat der Glycolyse.
Übersicht über den Aminozucker-Stoffwechsel
BearbeitenD-Fructose-6-P ⇓⇑ 2.6.1.16 / 3.5.99.6 D-Glucosamin-6-P ⇐ D-Glucosamin ⇐ D-Glucosaminid 2.7.1.1 3.2.1.- ⇓ 2.3.1.4 oder ⇓⇑ 3.5.1.25 N-Acetyl-D-Glucosamin-6-P ⇐ N-Acetyl-D-Glucosamin ( ⇐ Chitobiose ) ⇐ Chitin 2.7.1.59 3.2.1.52 3.2.1.14 ⇓⇑ 5.4.2.3 N-Acetyl-D-Glucosamin-1-P ⇓⇑ 5.1.3.8 ⇓⇑ 2.7.7.23 2.7.1.60 UDP-N-Acetyl-D-Glucosamin ⇔ N-Acetyl-D-Mannosamin ⇒ N-Acetyl-D-Mannosamin-6-P 5.1.3.14 ⇔ ⇓⇑ 5.1.3.7 ⇓⇑ 5.1.3.14 ⇓⇑ 2.5.1.56 3.1.3.- ⇓⇑ 2.5.1.56 / 2.5.1.57 UDP-N-Acetyl- UDP-N-Acetyl- N-Acetylneuraminat D-Galactosamin D-Mannosamin N-Acetylneuraminat-9-P ⇓⇑ 2.7.7.43 CMP-N-Acetylneuraminat
Einzelreaktionen
BearbeitenVom D-Fructose-6-phosphat zum UDP-N-Acetyl-D-Galactosamin und -Mannosamin
Bearbeiten| ⇓ | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | ||
|---|---|---|---|---|---|---|---|---|---|
|
|||||||||
| L-Glutamin | L-Glutamin | Glutamin--Fructose-6-phosphat- Transaminase (isomerisierend) | 2.6.1.16 | Tr | |||||
|
H2O |
oder |
H2O |
oder Glucosamin-6-phosphat-Deaminase |
3.5.99.6 | Hyd | ||||
|
|||||||||
| Acetyl-CoA | Glucosamin-6-phosphat- N-Acetyltransferase | 2.3.1.4 | Tr | ||||||
|
H2O |
oder |
H2O |
oder N-Acetylglucosamin-6-phosphat- Deacetylase |
3.5.1.25 | Hyd | ||||
|
|||||||||
| Phosphoacetylglucosamin- Mutase | 5.4.2.3 | Iso | |||||||
|
|||||||||
| UTP
PPi |
UTP
PPi |
UDP-N-Acetylglucosamin- Diphosphorylase | 2.7.7.23 | Tr | |||||
|
|||||||||
| UDP-N-Acetylglucosamin- 4-Epimerase | 5.1.3.7 | Iso | |||||||
|
|||||||||





| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | ||
|---|---|---|---|---|---|---|---|---|---|
|
|||||||||
| UDP-N-Acetylglucosamin- 2-Epimerase | 5.1.3.14 | Iso | Inclusion body- und Nonaka-Myopathie, Sialurie | ||||||
|
|||||||||

Abbau von D-Glucosaminid und D-Glucosamin
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | ||
|---|---|---|---|---|---|---|---|---|---|
|
|||||||||
| H2O
D-Glucosaminid |
? | 3.2.1.- | Hyd | ||||||
|
|||||||||
| ATP
ADP |
Hexokinase | 2.7.1.1 | Tr | Hämolyt. Anämie bei HK1-Def. | |||||
|
|||||||||

Abbau von Chitin und Chitobiose
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | ||
|---|---|---|---|---|---|---|---|---|---|
|
|||||||||
|
m H2O n N-Acetyl-D-Glucosamin, Chitin |
Chitinase | 3.2.1.14 | Hyd | ||||||
|
|||||||||
| H2O
N-Acetyl-D-Glucosamin |
β-N-Acetylhexosaminidase | 3.2.1.52 | Hyd | GM2-Gangliosidose I (Tay-Sachs), II (Sandhoff) | |||||
|
|||||||||
| ATP
ADP |
N-Acetylglucosamine-Kinase | 2.7.1.59 | Tr | ||||||
|
|||||||||


Chitin findet man in der Zellwand von Pilzen und im Außenskelett von Arthropoden, wo es für dessen Flexibilität verantwortlich ist. Strukturell entspricht es der pflanzlichen Zellulose (β-1,4-Bindungen), wobei statt D-Glucose das N-Acetyl-D-Glucosamin verwendet wird.
Biosynthese / Abbau von CMP-N-Acetylneuraminat
Bearbeiten| ⇓ | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | ||
|---|---|---|---|---|---|---|---|---|---|
|
|||||||||
| H2O
UDP |
H2O
UDP |
UDP-N-Acetyl- glucosamin- 2-Epimerase | 5.1.3.14 | Iso | Inclusion body- und Nonaka- Myopathie, Sialurie | ||||
|
|||||||||
| H2O, PEP
Pi |
PEP, H2O
PPi |
N-Acetylneuraminat- Synthase | 2.5.1.56 | Tr | |||||
|
|||||||||
| CTP
PPi |
CTP
PPi |
N-Acylneuraminat- Cytidylyltransferase | 2.7.7.43 | Tr | |||||
|
|||||||||

Biosynthese und Abbau von N-Acetylneuraminat-9-phosphat
Bearbeiten| ⇓ | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | ||
|---|---|---|---|---|---|---|---|---|---|
|
|||||||||
| ATP
ADP |
N-Acylmannosamin-Kinase | 2.7.1.60 | Tr | Inclusion body- und Nonaka- Myopathie, Sialurie | |||||
|
H2O |
oder |
H2O |
oder ? |
3.1.3.- | Hyd | ||||
|
|||||||||
| H2O, PEP
Pi |
PEP, H2O
PPi |
N-Acetylneuraminat-Synthase | 2.5.1.56 | Tr | |||||
| N-Acetylneuraminat-9-phosphat-Synthase | 2.5.1.57 | Tr | |||||||
|
|||||||||


N-Acetyl-D-Mannosamin kann zu N-Acetyl-D-Glucosamin isomerisiert werden
Bearbeiten| ⇓ | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | ||
|---|---|---|---|---|---|---|---|---|---|
|
|||||||||
| ATP | N-Acylglucosamin-2-Epimerase | 5.1.3.8 | Iso | ||||||
|
|||||||||
Allgemeines
BearbeitenAminozucker werden aus D-Fructose-6-phosphat gebildet. Aminozucker gehen nach Aktivierung durch CTP oder UTP beim Menschen in den Heteroglycan-Stoffwechsel ein, z.B. in die Synthese des Saccharid-Anteils von Glycolipiden, Glycoproteinen und Proteoglykanen (Glycosaminoglycane). Bsp.:
- UDP-N-Acetyl-D-Glucosamin - Synthese von Heparansulfat und GPI-Ankern
- UDP-N-Acetyl-D-Galactosamin - Synthese von Chondroitinsulfat.
- CMP-N-Acetylneuraminat (NANA) - Biosynthese der Lewis-Antigene Sialyl-Lea (Lacto-Serie) und Sialyl-Lex (Neo-Lacto-Serie), zu denen beispielsweise die Blutgruppen-Antigene des AB0-Systems gehören. NANA findet sich auch häufig am Ende der Oligosaccharidkette von Glykoproteinen z.B. von Plasmaproteinen, deren Abbau dadurch verhindert wird. NANA kann von Neuraminidasen abgespalten werden.
Weitere Funktionen:
- N-Acetyl-D-Glucosamin (GlcNAc) - Kovalente Modifikation von Proteinen an Serin- und Threonin-Resten durch die N-Acetyl-D-Glucosamin-Transferase.
Weblinks
Bearbeiten
Inositolphosphat-Stoffwechsel
BearbeitenAllgemeines
BearbeitenInositol ist ein zyklischer 6-wertiger Alkohol, der aus Glucose gebildet wird. Die einzelnen Hydroxyl-Gruppen können durch Phosphate ersetzt und sein. Das Inositol(phosphat) kann weiterhin mit dem Phosphoglycerid Phosphatidsäure verbunden sein.
Phosphatidyl-Inositolphosphate spielen eine wichtige Rolle in der intrazellulären Signaltransduktion. Hier ist insbesondere die Spaltung von 1-Phosphatidyl-D-myo-Inositol-4,5-bisphosphat durch die Phospholipase C in die sekundären Botenstoffe Inositol-1,4,5-trisphosphat und Diacylglycerin hervorzuheben. Dieser Prozess wird durch ein G-Protein (Gq/11) gesteuert.
1-Phosphatidyl-1D-myo-inositol ist weiterhin der Ausgangspunkt für die Biosynthese von Glycosyl-Phosphatidylinositol-Ankern (GPI-Anker).
Biosynthese und Abbau von Inositol
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
| NAD | Inositol-1-phosphat-Synthase | 5.5.1.4 | Iso | ||||
|
|||||||
| H2O
Pi |
Inositol-phosphat-Phosphatase | 3.1.3.25 | Hyd | ||||
|
|||||||
| O2
H2O |
Fe | Inositol-Oxygenase | 1.13.99.1 | Ox | |||
|
|||||||


D-Glucuronat fällt auch im Glucuronsäuren-Stoffwechsel an.
Biosynthese von 1-Phosphatidyl-D-myo-Inositol-4,5-bisphosphat (PI(4,5)P2)
BearbeitenEs gibt 2 Möglichkeiten für die Biosynthese von Phosphatidylinositol-4,5-bisphosphat:
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| CDP-Diacylglycerin | CDP-Diacylglycerin--Inositol- 3-phosphatidyltransferase | 2.7.8.11 | Tr | |||||||
|
||||||||||
| ATP
ADP |
1-Phosphatidylinositol-4-Kinase | 2.7.1.67 | Tr | |||||||
|
||||||||||
| ATP
ADP |
1. |
Pi
H2O |
1) 1-Phosphatidylinositol-4-phosphat-5-Kinase | 2.7.1.68 | Tr | |||||
| 2) Phosphoinositid- 5-Phosphatase | 3.1.3.36 | Hyd | Lowe-S., Dent- Krankheit 2 | |||||||
|
||||||||||



2. Möglichkeit:
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| CDP-Diacylglycerin | CDP-Diacylglycerin--Inositol- 3-phosphatidyltransferase | 2.7.8.11 | Tr | |||||||
|
||||||||||
| ATP
ADP |
1-Phosphatidylinositol-3-Kinase | 2.7.1.137 | Tr | |||||||
|
||||||||||
| 2 ATP
2 ADP |
Kinasen | ? | Tr | |||||||
|
||||||||||
| H2O
Pi |
1. |
ADP
ATP |
Mg | 1) Phosphatidylinositol- 3,4,5-trisphosphat-3-Phosphatase | 3.1.3.67 | Hyd | Bannayan-Riley- Ruvalcaba-S., Cowden-S., Makrozephalie- Autismus-S. | |||
| 2) Phosphatidylinositol- 4,5-bisphosphat-3-Kinase | 2.7.1.153 | Tr | ||||||||
|
||||||||||


Spaltung von PI(4,5)P2 in Diacylglycerin (DAG) und I(1,4,5)P3
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||
| H2O
|
Phospholipase C | 3.1.4.11 | Hyd | ||||||||
|
|||||||||||


Inositol-1,4,5-trisphosphat und Diacylglycerin sind die sekundären Botenstoffe des Gq/11 - IP3-Signalweges. Diese werden freigesetzt, wenn die Phospholipase C durch das G-Protein Gq/11 aktiviert wurde. Dieser Signalweg ist neben dem ebenfalls G-Protein-gekoppelten Adenylatcyclase-Weg (mit dem sekundären Botenstoff cAMP) einer der häufigsten Signalübertragungswege, die ein extrazelluläres Signal (Rezeptoraktivierung) in ein intrazelluläres übersetzen.
Weiteres Schicksal von I(1,4,5)P3
BearbeitenD-myo-Inositol-1,4,5-trisphosphat wird auf verschiedenen Wegen durch Kinasen und Phosphatasen umgewandelt und z.B. in 1D-myo-Inositol überführt, das wie oben dargestellt zu Glucuronat oxidiert wird.
Bsp. 1:
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| H2O
Pi |
Inositol-polyphosphat-5-Phosphatase | 3.1.3.56 | Hyd | |||||||
|
||||||||||
| Pi
align="right"|H2O |
Inositol-1,4-bisphosphat-1-Phosphatase | 3.1.3.57 | Hyd | |||||||
|
||||||||||
| H2O
Pi |
Inositol-phosphat-Phosphatase | 3.1.3.25 | Hyd | |||||||
|
||||||||||


Bsp. 2:
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| ATP
ADP |
1. |
Pi
H2O |
Ca | 1) Inositol-trisphosphat-3-Kinase | 2.7.1.127 | Tr | ||||
| 2) Multiple-Inositol-polyphosphat-Phosphatase | 3.1.3.62 | Hyd | ||||||||
|
||||||||||
| H2O
Pi |
1. |
ADP
ATP |
1) Inositol-polyphosphat-5-Phosphatase | 3.1.3.56 | Hyd | |||||
| 2) Inositol-tetrakisphosphat-1-Kinase | 2.7.1.134 | Tr | ||||||||
|
||||||||||
| H2O
Pi |
Inositol-polyphosphat-1-Phosphatase | 3.1.3.57 | Hyd | |||||||
|
||||||||||
| H2O
Pi |
Phosphatidylinositol-3,4-bisphosphat-4-Phosphatase | 3.1.3.66 | Hyd | |||||||
|
||||||||||
| H2O
Pi |
Inositol-phosphat-Phosphatase | 3.1.3.25 | Hyd | |||||||
|
||||||||||




Bsp. 3:
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| ATP
ADP |
Inositol-polyphosphat-Multikinase | 2.7.1.151 | Tr | |||||||
|
||||||||||
| ATP
ADP |
Inositol-polyphosphat-Multikinase | 2.7.1.151 | Tr | |||||||
|
||||||||||
| ATP
ADP |
Inositol-polyphosphat-Multikinase | 2.7.1.- | Tr | |||||||
|
||||||||||



Glycosyl-Phosphatidylinositol-Ankern (GPI-Anker)
BearbeitenSiehe unter Glycosyl-Phosphatidylinositol-Anker (Abschnitt Glycane). Ausgangsstoff ist das 1-Phosphatidyl-D-myo-inositol.
Weblinks
Bearbeiten- KEGG: Inositol phosphate metabolism - Homo sapiens (human)
- KEGG: Glycosylphosphatidylinositol(GPI)-anchor biosynthesis - Homo sapiens (human)
Alkohol-Stoffwechsel
BearbeitenAbbau von Ethanol
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||
| NAD(P)+ | NAD(P)+ | Zn / Fe | Alkohol-Dehydrogenase (NAD+) | 1.1.1.1 | Ox | ||||||
| Zn | Alkohol-Dehydrogenase (NADP+) | 1.1.1.2 | |||||||||
|
|||||||||||
| H2O, NAD(P)+ | NAD(P)+, H2O | Aldehyddehydrogenase (NAD+) (Zytosol: ALDH1 , Mitochondrium: ALDH2) | 1.2.1.3 | Ox | ALDH2-Def. (Alkohol- intoleranz), ALDH3A2-Def. (Sjögren-Larsson-S.) | ||||||
| Aldehyddehydrogenase (NADP+) | 1.2.1.4 | ||||||||||
|
|||||||||||
| CoA-SH, ATP
AMP + PPi |
CoA-SH, ATP
AMP + PPi |
Acetat-CoA-Ligase | 6.2.1.1 | Lig | |||||||
|
|||||||||||
Ethanol entsteht in geringen Mengen z.B. durch Darmbakterien, im Intermediärstoffwechsel und wird in variabler Menge exogen aufgenommen. Der Abbau erfolgt wie oben dargestellt durch Oxidation des Alkohols zum Aldehyd und dann zur Carbonsäure. Letztere kann nach Aktivierung mit Coenzym A zur Energiegewinnung (Citratzyklus) oder für Biosynthesen (z.B. von Fettsäuren) verwendet werden. Der Ethanol-Abbau erfolgt in kleinerem Ausmaß auch über das CYP-abhängige mikrosomale Ethanol-oxidierende System (MEOS) der Leber.
Pharmakologie und Toxikologie: Toxisch ist neben dem Ethanol v.a. das Acetaldehyd.
Die Induktion des MEOS bei Alkoholismus und dessen Hemmung bei akuter Alkoholzufuhr führt zu pharmakokinetischen Wechselwirkungen.
Verstoffwechselung von Methanol
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||
| NAD(P)+ | NAD(P)+ | Zn / Fe | Alkohol-Dehydrogenase (NAD+) | 1.1.1.1 | Ox | ||||||
| Zn | Alkohol-Dehydrogenase (NADP+) | 1.1.1.2 | |||||||||
|
|||||||||||
| H2O, NAD(P)+ | NAD(P)+, H2O | Aldehyddehydrogenase (NAD+) (Zytosol: ALDH1 , Mitochondrium: ALDH2) | 1.2.1.3 | Ox | ALDH2-Def. (Alkohol- intoleranz), ALDH3A2-Def. (Sjoegren-Larsson-S., SLS) | ||||||
| Aldehyddehydrogenase (NADP+) | 1.2.1.4 | ||||||||||
|
|||||||||||
| THF, ATP
ADP, Pi |
Formiat--THF-Ligase | 6.3.4.3 | Lig | ||||||||
| 10-Formyl-THF | |||||||||||
Formaldehyd entsteht in geringen Mengen z.B. beim Cholin-Abbau. Ameisensäure (Formiat) wird z.B. bei der Cholesterin-Biosynthese, bei der Östrogen-Bildung, beim Abbau von Tryptophan und bei der Biosynthese von Biopterin frei. Formiat wird von der Formiat--THF-Ligase an Tetrahydrofolsäure gebunden, so dass 10-Formyl-THF entsteht, das dann weiter verstoffwechselt wird.
Pharmakologie und Toxikologie: Beim Abbau von Methanol entsteht Formaldehyd und Ameisensäure. Letzteres wird sehr langsam abgebaut, führt zur metabolischen Azidose und schädigt insbesondere die Sehnerven bis hin zur Erblindung. Die Vergiftung verläuft in 3 Phasen: Trunkenheit (schwächer als beim Ethanol), asymptomatische Latenzphase, schwere Vergiftung. Eine therapeutische Option ist die kontinuierliche Gabe von Ethanol in hoher Dosierung, um die Methanolverstoffwechselung durch die Enzyme kompetitiv zu hemmen.
Weblinks
Bearbeiten- KEGG: Glycolysis / Gluconeogenesis - Homo sapiens (human)
- KEGG: Fatty acid metabolism - Homo sapiens (human)
- RCSB PDB: Alcohol Dehydrogenase
Oxidative Phosphorylierung
BearbeitenDie Atmungskette
Bearbeiten.jpg) |
 |
 |
Allgemeines
BearbeitenAtmungskette und oxidative Phosphorylierung sind in den Mitochondrien, den „Kraftwerken der Zelle“, lokalisiert. Ihr Sinn besteht darin, die in den katabolen oxidativen Stoffwechselwegen gewonnene Energie in Form der Reduktionsäquivalente (NADH, FADH2) auf einen Energieträger zu übertragen, den die Zelle vielfältiger und flexibler nutzen kann: Das ATP.
Zuerst wird die Energie der Elektronen genutzt, um Protonen über die innere Mitochondrienmembran in den Membranzwischenraum zu transportieren, so dass an der inneren Mitochondrienmembran ein elektrochemischer Gradient aufgebaut wird. Der davon angetriebene Protonen-Rückstrom durch die ATP-Synthase (Komplex V) liefert dann die Energie, um aus ADP und anorganischem Phosphat die „universale Energiewährung“ der Zelle, ATP, zu erzeugen. Die Elektronen werden am Ende auf Sauerstoff übertragen und reagieren mit H+ zu Wasser: 2 H+ + 2 e− (= H2) + 1/2 O2 → H2O. Dies entspricht formal der explosiven Knallgasreaktion. Bei letzterer verpufft die ganze Energie jedoch in Form von Wärme. Erst die schrittweise Freisetzung der Energie in der Atmungskette erlaubt es der Zelle, die Energie zu nutzen und in Form eines Protonengradienten bzw. einer chemischen Bindung zu speichern.
Aus dem Gesagten ergibt sich auch, dass der Sauerstoff, den wir einatmen, sich nicht im Kohlendioxid befindet, das wir ausatmen, sondern in den etwa 300 ml Oxidationswasser, die wir pro Tag produzieren.
Die Komplexe I bis IV
BearbeitenDie unter anderem im Citratzyklus und in der β-Oxidation gewonnenen Reduktionsäquivalente (NADH) können ihre energiereichen Elektronen auf die Komplexe der Atmungskette übertragen. Neben den Flavoproteinen FAD und FMN spielen sog. Eisen-Schwefel-Cluster und Häm-Moleküle eine große Rolle bei der Aufnahme und Weitergabe der Elektronen.
Der Elektronentransport zwischen den Komplexen I bis IV wird bewerkstelligt vom Ubichinon (Coenzym Q) und dem Häm-haltigen Cytochrom c. Q überträgt die Elektronen von den Dehydrogenase-Komplexen I und II auf Komplex III. Cytochrom c überträgt jeweils ein Elektron vom Komplex III auf Komplex IV.
- Komplex I (NADH-Dehydrogenase, EC 1.6.99.3) - Der FMN-haltige Komplex I kann Elektronen von NADH übernehmen, pumpt damit H+ über die innere Mitochondrienmembran und gibt sie dann über Q an den Komplex III weiter. Die NADH-Dehydrogenase enthält funktionell bedeutsame Eisen-Schwefel-Zentren.
- Komplex II (Succinat-Dehydrogenase, EC 1.3.99.1) - Die Succinat-Dehydrogenase ist ein Enzym des Citratzyklus mit dem Kofaktor FAD. FAD oxidiert Succinat zu Fumarat und gibt die aufgenommenen Elektronen über Q ebenfalls an den Komplex III weiter, jedoch ohne Protonen über die Membran zu pumpen. Die Succinat-Dehydrogenase enthält weiterhin Eisen-Schwefel-Zentren und Häm-Moleküle.
- Komplex III (Cytochrom-c-Reduktase, Cytochrom-bc1-Komplex, EC 1.10.2.2). Der Cytochrom bc1-Komplex übernimmt die Elektronen vom reduzierten Q (Ubichinol, QH2), transportiert damit Protonen und gibt sie einzeln an Cytochrom c weiter. Auch die Cytochrom-c-Reduktase enthält Eisen-Schwefel-Cluster und Häm-Moleküle.
- Komplex IV (Cytochrom-c-Oxidase, EC 1.9.3.1) - Die Cytochrom-c-Oxidase übernimmt die Elektronen, pumpt noch einmal Protonen, bevor sie letztlich mit Sauerstoff und Protonen Wasser bilden (Sauerstoff wird zu Wasser reduziert). Sie enthält ebenfalls Häm-Moleküle.
Die ATP-Synthase (Komplex V)
BearbeitenDer aufgebaute Protonengradient wird nun genutzt, um elektro-mechanisch die Protonenturbine (F0-Motor in der Membran) der ATP-Synthase anzutreiben. Der dadurch in Drehung versetzte Rotor ist über eine Achse mit einem zweiten Motor, dem F1-Motor verbunden, der dadurch zum Generator wird. Die Achse verschiebt die Untereinheiten des F1-Teils in rhythmischer Folge so gegeneinander, dass in seinen Bindungstaschen aus ADP und Pi ATP „gepresst“ werden kann. Von einem Stator werden die Teile zusammengehalten.
Die ATP-Synthase ist quasi eine Protonenpumpe, die rückwärts läuft (Eine Protonenpumpe pumpt H+ unter ATP-Verbrauch). Die ATP-Synthase kann tatsächlich auch rückwärts laufen und unter ATP-Verbrauch Protonen pumpen, allerdings nicht sehr effizient.
Entkopplung der Atmungskette
BearbeitenZurückfließende Protonen, die nicht zur ATP-Synthese genutzt werden setzen die freie Enthalpie, die im Gradienten gespeichert ist als Wärme frei. Die plurivakuolären Zellen des braunen Fettgewebes, das sich u.a. bei Winterschläfern und bei menschlichen Säuglingen (am Rücken und entlang der großen Gefäße) findet nutzen diesen Mechanismus zur effektiven Wärmeerzeugung. Das verantwortliche Protonenkanalprotein ist das Thermogenin. Stimuliert wird dieser Prozess über β3-Adrenozeptoren (cAMP↑), so dass die Lipolyse gesteigert und die Biosynthese von Thermogenin und Lipoproteinlipase induziert wird.
Pathologie
BearbeitenVerschiedene angeborene Defekte der Atmungsketten-Enzyme können das neurodegenerative Leigh-Syndrom verursachen (OMIM).
Toxikologie
BearbeitenDinitrophenol (DNP), ein Stoff aus der Sprengstoffherstellung ist ein chemischer Entkoppler. Das Molekül bewegt sich frei in der inneren Mitochondrienmembran und kann Protonen auf der einen Seite aufnehmen und auf der anderen Seite wieder abgeben. Als Diätmittel ist DNP effektiv, aber leider mitunter tödlich. Die Wirkungsweise führt zur Hyperthermie („Dieting by cooking yourself“), zum ATP-Defizit und durch die Unterbindung des aeroben Stoffwechsels zu Gunsten des anaeroben Glucoseabbaus zur metabolischen Azidose. Zudem ist DNP krebserregend.
Cyanide (-C≡N) wie z.B. Blausäure (HCN) oder Zyankali (KCN) hemmen die Cytochrom-c-Oxidase (Komplex IV) der Atmungskette und führen dadurch zur inneren Erstickung.
| Komplex | Hemmstoffe |
| I (NADH:Q-Oxidoreduktase) | Rotenon, Amytal (ein Barbiturat) |
| III (Q:Cytochrom c-Oxidoreduktase) | Antimycin A, Stigmatellin, Myxothiazol |
| IV (Cytochrom c:O2-Oxidoreduktase) | Azide, Cyanide, Kohlenmonoxid |
| V (ATP-Synthase) | Oligomycin, DCC (Dicyclohexylcarbodiimid) |
Sauerstoff - Dr. Jekyll und Mr. Hyde
BearbeitenSauerstoff ist als Endakzeptor der Elektronen, die beim Abbau organischer Moleküle gewonnen und in der Atmungskette ihrer Energie beraubt werden, für alle Eukaryonten und viele Bakterien (Aerobier) lebensnotwendig. Sauerstoffentzug führt binnen kürzester Zeit durch Sistieren der Energieproduktion zum Tod.
Auf der anderen Seite gehen durch Fehlübertragungen von Elektronen in der Atmungskette oder bei anderen Redoxvorgängen wie z.B. an den P450-Cytochromen (Phase I der Biotransformation in Leber und Darmmucosa) aus dem Sauerstoff sog. reaktive Sauerstoffspezies (reactive oxygen species, ROS) hervor, die aufgrund ihrer Reaktionsfreudigkeit Zellbestandteile wie DNA, Proteine und Membranlipide schädigen können. Dies wird vom Körper auch genutzt, indem z.B. Granulozyten mit Hilfe der NAD(P)H-Oxidase (Co: Ca, FAD, Häm. EC 1.6.3.1) im Rahmen der Entzündungsreaktion ROS zur Abwehr von Bakterien bilden. ROS können durch Bildung weiterer Radikale z.B. dem Lipidperoxylradikal LOO˙ und dem Alkoxylradikal LO˙ richtige Kettenreaktionen in Gang setzen, die z.B. zur ausgiebigen Lipidperoxidation von Zellmembranen führen können. Dabei sind v.a. ungesättigte Fettsäuren das Ziel oxidativer Angriffe. Aus diesem Grund verfügt die Zelle über ein ganzes Arsenal an Abwehrmechanismen gegenüber dem alltäglichen „oxidativen Stress“. Dazu gehört:
- Das Antioxidans Vitamin C (Ascorbat) in Kooperation mit Vitamin E (Tocopherol).
- Das Glutathion-System, das vor allem in Leber und Erythrozyten wichtig ist.
- Die Enzyme Superoxiddismutase (SOD) und Katalase.
Auf letztere soll an dieser Stelle kurz eingegangen werden. Die Superoxiddismutase (SOD) (Co: Fe, Mn oder Cu und Zn. EC 1.15.1.1) entgiftet das reaktive Superoxid (Superoxidanionradikal) O2˙- durch Disproportionierung von zwei Superoxidanionradikalen mit Hilfe von zwei Wasserstoffionen (Protonen) H+ zu Wasserstoffperoxid H2O2 und Sauerstoff O2:

 |
Das ebenfalls reaktive Wasserstoffperoxid H2O2 wird im nächsten Schritt durch die Katalase (Co: Häm, Mn. EC 1.11.1.6) zu Wasser und Sauerstoff disproportioniert:

Pathologie
Bearbeiten- ROS stellen einen wichtigen Schädigungsmechanismus dar, sowohl bei akuten Stoffwechselentgleisungen als auch bei chronischer Zellschädigung und Alterungsprozessen.
- 15 bis 20 % der Fälle von familiärer amyotropher Lateralsklerose (FALS, ALS1), einer neurodegenerativen Erkrankung, sind mit Mutationen im Superoxiddismutase-1-Gen (SOD1, Chromosom 21q22.1) assoziiert (OMIM).
- Ein Defekt des NADPH-Oxidase-Komplexes in den Granulozyten führt zur chronischen Granulomatose (OMIM, OMIM, OMIM, OMIM) bzw. zum neutrophilen Immundefizienz-Syndrom (OMIM).
Weblinks
BearbeitenAllgemein:
- KEGG: Atmungskette und ATP-Synthase
- Atmungskette
- Die Zellatmung zum Ausfüllen
- Mécanisme de synthèse d'ATP
- ATP Synthase: eine prächtige molekulare Maschine
RCSB PDB:
- RCSB PDB: Complex I
- RCSB PDB: Cytochrome bc1
- RCSB PDB: Cytochrome c
- RCSB PDB: Cytochrome c Oxidase
- RCSB PDB: ATP Synthase
- RCSB PDB: Superoxide Dismutase
- RCSB PDB: Catalase
Animationen:
- YouTube: The Dynamic Cell
- YouTube: F1 ATP synthase
- YouTube: ATP synthase - top view
- YouTube: Rotation of the gamma subunit effects ATP synthesis
- YouTube: Conformational Changes in the Beta Subunits of the ATPase
- YouTube: Molecular Mechanism of ATP synthesis
- Animation Movies of ATP Synthase
- Elektronentransportkette
- ATP-Synthase
- Atemkette
- ATP-Synthase
- Mitochondrialer Elektronentransport
- Atmungskette
- Atmungskette
- ATP-Synthase
- ATP-Synthese-Mechanismus
- ATP-Synthase
- Rotation des F0-Motors
Direktbeobachtung:
- F1-ATPase: A Rotary Motor Made of a Single Molecule
- Load Dependence of F1 Rotation
- Stepping Rotation of F1-ATPase at Low ATP Concentrations
- Stepping Rotation of F1-ATPase Revealed by Single-Fluorophore Imaging
- Full-Speed Rotation of F1-ATPase
- Substeps in F1 Rotation
Fun:
Porphyrinbiosynthese
BearbeitenAllgemeines
BearbeitenDie farbigen Häm-Moleküle dienen im Blut in Form des Hämoglobins als Sauerstofftransportmittel, im Muskel als Sauerstoffspeicher. Häm dient weiterhin vielen Enzymen, die Redoxreaktionen und Elektronentransfers ermöglichen, als prosthetische Gruppe.
Biosynthese eines Häm aus 8 Succinyl-CoA und 8 Glycin
Bearbeiten| Tr. | Tl. | Lok. | All. | ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC/EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||||
| - Häm | - Häm (HRE) | - Häm | Pyrid- oxal- phos- phat | δ-Aminolävulinat- Synthase 1 (δ-ALA-S1) Lebermitochondrien | 2.3.1.37 Tr | ||||||||
| + EPO | - Fe- Mangel (IRP1) | - Häm (HRE) | δ-Aminolävulinat- Synthase 2 (δ-ALA-S2) Knochenmitochondrien | Sideroblastische Anämie (XLSA) | |||||||||
|
|||||||||||||
|
|
spontan | ||||||||||||
|
|||||||||||||
|
|
Zn | Porphobilinogen- Synthase (δ-ALA- Dehydratase)
Zytosol |
4.2.1.24 Ly | δ-ALA- Dehydratase- Mangel (ADM), Akute hepatische Porphyrie | |||||||||
|
|||||||||||||
| H2O
|
Dipyrro- methan | PBG-Deaminase
Zytosol |
2.5.1.61 Tr | Akute intermittierende Porphyrie (AIP) | |||||||||
|
|||||||||||||
|
|
Uroporphyrinogen-III- Synthase (Isomerase)
Zytosol |
4.2.1.75 Ly | Kongenitale erythropoetische Porphyrie (KEP) | ||||||||||
|
|||||||||||||
|
|
Uroporphyrinogen- Decarboxylase
Zytosol |
4.1.1.37 Ly | Porphyria cutanea tarda (PCT), Hepatoerythro- poetische Porphyrie (HPP) | ||||||||||
|
|||||||||||||
| O2
2 H2O, 2 CO2 |
Koproporphyrinogen- Oxidase
Mitochondrium |
1.3.3.3 Ox | Hereditäre Koproporphyrie (HKP) | ||||||||||
|
|||||||||||||
|
|
FAD | Protoporphyrinogen- Oxidase
Mitochondrium |
1.3.3.4 Ox | Porphyria variegata (PV) | |||||||||
|
|||||||||||||
| + EPO | Fe2+
2 H |
Ferrochelatase
Mitochondrium |
4.99.1.1 Ly | Protoporphyrie (PP) | |||||||||
|
|||||||||||||








Die Biosynthese von Häm aus Succinyl-CoA und Glycin umfasst acht enzymatisch katalysierte Schritte und verteilt sich auf zwei zelluläre Kompartimente: Zytosol und Mitochondrium. Die Regulation erfolgt hauptsächlich auf Höhe der δ-Aminolävulinat-Synthase.
Eigenschaften und biologische Bedeutung
BearbeitenPorphyrine (πορφυρά, porphyrá (griech.): Purpurfarbstoff) bestehen aus vier Pyrrol-Ringen, die durch vier Methin-Gruppen kreisförmig verbunden sind. Die Farbigkeit vieler Porphyrine geht auf die konjugierten Doppelbindungen und aromatischen Eigenschaften zurück. Neben dem intensiv roten Häm, das dem Blut seine Farbe gibt, gehört z.B. auch das grüne Chlorophyll der Pflanzen dazu. Für die Funktion ist die Komplexierung eines Metallions im Ringsystem essentiell. Beim Häm ist dies Eisen, beim Chlorophyll Magnesium. Die fünfte Koordinationsstelle des Häm-Eisens wird vom Stickstoff einer Histidin-Seitenkette des Hämoglobins besetzt. Vom Biosyntheseweg der Porpyrine zweigt auch der Bildungsweg der verwandten Cobalamine (Vitamin B12) ab. Diese werden nur von Mikroorganismen gebildet und bestehen aus einem Corrin-Ringsystem, das zentral ein Kobaltion komplexiert. Gemeinsam ist allen, dass sie vier verbundene Pyrrole (ein Pyrrol ist ein aromatischer 5-Ring aus vier Kohlenstoff und einem Stickstoff) enthalten und damit ein Tetrapyrrol bilden.
Die Häm-Synthese findet in allen Körperzellen statt, da Häm für die Atmungskette benötigt wird, eine besonders hohe Syntheserate findet sich jedoch in den erythropoetischen Vorläuferzellen im Knochenmark (Hämoglobin-Bildung) sowie in der Leber (Biotransformation Phase I).
Das zentrale Eisenion im Häm kann leicht Elektronen aufnehmen und abgeben, d.h. zum Fe3+ oxidiert und zum Fe2+ reduziert werden. Daher werden Hämmoleküle v.a. bei Oxidoreduktasereaktionen eingesetzt, z.B. bei der Entgiftung (Disproportionierung) von Wasserstoffperoxid (H2O2) zu Sauerstoff (O2) und Wasser (H2O) (Katalase (Häm b)), im Rahmen der hepatischen Biotransformation Phase I (Cytochrom P450 Oxidase (Häm b)), in der Cyclooxygenase-Reaktion der Prostaglandinbiosynthese, (Cyclooxygenase (Häm b)) und in der mitochondrialen Elektronentransportkette (Succinatdehydrogenase (Häm b), Cytochrom c Reduktase (2x Häm b, Häm c), Cytochrom c Oxidase (2x Häm a) und Cytochrom c Peroxidase (Häm b)). Der Häm-Fe2+-Komplex kann an der sechsten freien Koordinationsstelle auch leicht reversibel molekularen Sauerstoff binden und wird daher als Sauerstofftransportmittel (Hämoglobin (Häm b)) und -speicher (Myoglobin (Häm b)) verwendet.
Der Transkription des ersten (δ-ALA 2 der Erythroblasten) und des letzten Häm-Biosyntheseenzyms wird von Erythropoetin (EPO) induziert. Das Glycoprotein-Hormon wird bei peripherem Sauerstoffmangel (z.B. bei Anämie, chronischen Lungenerkrankungen, Herzinsuffizienz, Höhenaufenthalte) von der Niere freigesetzt und stimuliert nicht nur die Hämbiosynthese, sondern auch die Reifung der roten Vorläuferzellen.
Weiterhin wird die Hämbiosynthese an das verfügbare Eisen und das bereits vorhandene Häm angepasst, hier spielen das Eisen-sensorische und das Häm-regulatorische Element ein Rolle.
In der Leber wird Häm sowohl für die Cytochrome der Atmungskette benötigt als auch als Cytochrom P450 für die Biotransformation. Die Startreaktion der Hämbiosynthese wird in den Leberzellen zusätzlich transkriptionell und allosterisch durch Häm gehemmt, so dass die Synthese streng nach Bedarf aktiviert wird.
Pathobiochemie
BearbeitenEnzymdefekte führen zum Krankheitsspektrum der Porphyrien.
Pharmakologie
BearbeitenBei renaler Anämie (Niereninsuffizienz) muss Erythropoetin (EPO), das normalerweise von der Niere gebildet wird, substituiert werden. EPO wird auch als Dopingmittel missbraucht, um den Hb und damit die Sauerstofftransportkapazität des Blutes zu erhöhen.
Toxikologie
BearbeitenBlei ersetzt das Zinkion im aktiven Zentrum der Porphobilinogensynthase (δ-ALA-Dehydratase), die dadurch funktionsuntüchtig wird. Auch die Funktion der Koproporphyrinogen-Oxidase wird beeinträchtigt. In der Folge kommt es zum Anstau von δ-Aminolävulinat und zur Anämie. Auch der Coproporphyrinogen III-Spiegel ist erhöht. Blei führt weiterhin zur Neuropathie (z.B. N. radialis-Lähmung). Blei wird vor allem im Knochen abgelagert. Bleiexposition in der Schwangerschaft führt zur mentalen Retardierung.
Kohlenmonoxid (CO) bindet mit einer 200- bis 300-fach höheren Affinität an das Häm im Hämoglobin als Sauerstoff und kann daher bereits bei niedriger Luftkonzentration zu hohen CO-Hb-Spiegeln führen, die am Sauerstofftransport nicht mehr teilnehmen können. Die CO-Bindung führt außerdem zu einer Linksverschiebung der Sauerstoffbindungskurve, so dass im peripheren Gewebe auch die Sauerstoffabgabe des verbliebenen Oxy-Hämoglobins erschwert wird. Es kommt rasch zum inneren Ersticken. CO entsteht bei unvollständiger Verbrennung und wird besonders beim Betrieb von Heizungen und Öfen in schlecht gelüfteten Räumen zur Gefahr.
Cyanide (-C≡N) wie z.B. Blausäure (HCN) oder Zyankali (KCN) hemmen die Cytochrom c Oxidase (Komplex IV) der Atmungskette und führen dadurch zur inneren Erstickung.
Wird das Fe2+ im Hämoglobin zum Fe3+ oxidiert, so entsteht das braune Methämoglobin (MetHb, Hämiglobin). Dieses kann Sauerstoff zwar noch gut binden, aber peripher nur noch schwer abgeben. Eine Methämoglobinämie kann so zum inneren Ersticken führen, obwohl die pulsoxymetrisch messbare Sauerstoffsättigung völlig normal ist. Methämoglobin entsteht physiologisch durch Autooxidation, was durch die Methämoglobinreduktase ständig rückgängig gemacht wird. Die normale MetHb-Konzentration liegt unter 1 %. Eine Methämoglobinämie kann hervorgerufen werden durch 1) Vergiftungen mit Oxidationsmitteln (Chlorate), Nitrite, Amylnitrit, Nitroglyzerin, aromatische Amino- und Nitroverbindungen (Anilin, Nitrobenzol), 2) Enzymdefekte (Methämoglobin-Reduktase-Defizienz, Glucose-6-Phosphat-Dehydrogenase-Mangel) und genetische Störungen des Hämoglobins (Hämoglobin M) und 3) durch manche Arzneimittel bes. bei den vorgenannten Erkrankungen. Hierzu gehören Lokalanästhetika (Benzocain, Lidocain, Procain), Malariamittel (Primaquin), Sulfonamide und Paracetamol sowie Phenazopyridin.
Weblinks
Bearbeiten
Porphyrinabbau
BearbeitenAllgemeines
BearbeitenBeim Porphyrin-Abbau wird Häm zu Bilirubin und anderen Gallenfarbstoffen degradiert. Die Ausscheidung von Bilirubin erfolgt hauptsächlich über die Leber nach erfolgter Glucuronidierung.
Abbau der Porphyrine
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||
| 3 O2, 3 NADPH/H+
Fe2+, CO, 3 H2O, 3 NADP+ |
Häm-Oxygenase
RES |
1.14.99.3 | Ox | HMOX1-Def. | ||||||||
|
||||||||||||
| NADPH/H+ | Biliverdin-Reduktase
RES |
1.3.1.24 | Ox | |||||||||
|
||||||||||||
| UDP-Glucuronat
UDP |
Glucuronosyltransferase
Hepatozyt |
2.4.1.17 | Tr | Hyperbilirubinämie I (Gilbert-S.), Crigler-Najjar-S. Typ I und II, | ||||||||
|
||||||||||||
| Bilirubinmonoglucuronosid
Bilirubin |
Bilirubin-Glucuronosid- Glucuronosyltransferase
Hepatozyt |
2.4.1.95 | Tr | |||||||||
|
||||||||||||
| 2 H2O
2 Glucuronat |
β-Glucuronidase
Darm |
3.2.1.31 | Hyd | Mucopoly- saccharidose VII | ||||||||
|
||||||||||||
| 6 H
|
Darm | |||||||||||
|
||||||||||||
| 2 H
|
Darm | |||||||||||
|
||||||||||||
| 4 H
|
Darm | |||||||||||
|
||||||||||||










Beim Abbau von Erythrozyten im retikuloendothelialen System von Milz und Lebersinusoiden wird Hämoglobin freigesetzt. Freies Hämoglobin wird von Haptoglobin gebunden. Das Hämoglobin wird in Globin und Häm zerlegt. Häm wird von der Häm-Oxygenase unter Freisetzung des Eisens und Abspaltung von Kohlenmonoxid zum grünlichen Biliverdin oxidiert. Die Biliverdin-Reduktase reduziert Biliverdin weiter zum rot-gelb-bräunlichen Bilirubin. Bilirubin wird von der Leber aus dem Blut aufgenommen, und da es schlecht wasserlöslich ist, mit Glucuronsäure konjugiert und in die Gallenkanälchen sezerniert. Im Darm wird es teilweise dekonjugiert und von Bakterien weiter abgebaut. Die Gallenfarbstoffe werden z.T. reabsorbiert und über die Nieren ausgeschieden. Sie sind für die braune Farbe des Stuhls (Stercobilin) und die gelbe Farbe des Urins (Urobilinogen, Urobilin) verantwortlich.
Pathobiochemie
BearbeitenEin Anstieg des gelbrotbräunlichen Bilirubins im Blut durch vermehrte Produktion oder gestörte Ausscheidung führt zum Ikterus (Gelbsucht) mit gelber Verfärbung der Skleren und der Haut. Die Ursachen können prähepatisch (Hämolyse, z.B. bei Malaria), intrahepatisch (z.B. bei Leberzirrhose, Gilbert- oder Crigler-Najjar-Syndrom) oder posthepatisch (Cholestase, z.B. bei Gallensteinen) zu finden sein. Das erhöhte Serum-Bilirubin wird vermehrt über die Niere ausgeschieden und führt klassischerweise zum „bierbraunen Urin mit gelbem Schüttelschaum“.
Labormedizin
BearbeitenBeim prähepatischen Ikterus ist vor allem das unkonjugierte (indirekte) Bilirubin erhöht, beim intra- und posthepatischen Ikterus eher das konjugierte (direkte) Bilirubin.
Weblinks
Bearbeiten
Lipid-Stoffwechsel
BearbeitenFunktionen der Lipide und Steroide
Bearbeiten- Bausubstanz der Zellmembranen (Phospholipide, Cholesterin).
- Bausubstanz des Körpers (Baufett aus Fettzellen, die intrazellulär Fette speichern, z.B. retroperitoneal oder orbital-retrobulbär als Lückenfüller, sowie subkutan zur Wärmeisolierung).
- Energiegewinnung und Energiereserve besonders für längere Fastenzeiten (Speicherfett).
- Rohstoffe für Signalmoleküle:
- Cholesterin: Biosynthese von Steroidhormonen wie Mineralkortikosteroiden (Aldosteron) in der Zona glomerulosa der NNR, Glukokortikosteroiden (Cortisol) in der Zona fasciculata der NNR und Sexualsteroidhormonen (Androgene, Östrogene, Progesteron) in der Zona reticularis der NNR und in den Gonaden.
- Arachidonsäure: Rohstoff für die Biosynthese von Prostaglandinen und Leukotrienen, die als Entzündungsmediatoren fungieren.
- 1-Phosphatidyl-Inositol-4,5-bisphosphat: Die Phospholipase C setzt daraus die intrazellulären sekundären Botenstoffe Inositol-1,4,5-trisphosphat (IP3) und Diacylglycerin (DAG) frei.
Resorption und Verteilung
BearbeitenTriglyceride werden im Dünndarm nach Emulgierung durch die Gallensäuren v.a durch die Pankreas-Lipase hydrolytisch in Fettsäuren und β-Monoacylglycerin gespalten. Diese werden zusammen mit Cholesterin, anderen Lipiden, fettlöslichen Vitaminen und Pharmaka nach Bildung von Micellen resorbiert.
Im Dünndarmepithel erfolgt die Bildung von Apolipoprotein B48-haltigen Chylomikronen, die über den intestinalen Lymphstrom (Ductus thoracicus) abtransportiert werden und im linken Venenwinkel ins Blut gelangen. Nach fettreichen Mahlzeiten kann die hohe Konzentration von Chylomikronen zu einer Trübung des Blutplasmas führen. In der Leber werden die Lipide durch eine Lipoproteinlipase freigesetzt und in andere Lipoproteine eingebaut (VLDL, LDL, HDL). Übrig bleiben die Chylomikron-Restkörperchen (Remnants).
Der Lipidtransport von der Leber zur Peripherie erfolgt insbesondere über LDL, der Transport von der Peripherie zur Leber über HDL.
VLDL enthalten vor allem Triglyceride. Der Proteinanteil enthält das Apolipoprotein B100. Mit Hilfe von Apolipoprotein CII aktivieren VLDL die Endothel-ständige Lipoproteinlipase. Dadurch werden Fettsäuren freigesetzt und die VLDL zu IDL abgebaut.
LDL sind besonders cholesterinreich. Der Proteinbestandteil besteht überwiegend aus Apolipoprotein B100. Dieses wird in der Peripherie von Apolipoprotein-Rezeptoren erkannt, das LDL aufgenommen und lysosomal abgebaut. Das in der Zelle freigesetzte Cholesterin hemmt dann die zelleigene Cholesterin-Biosynthese.
Auf den HDL findet sich in hoher Aktivität die Lecithin-Cholesterin-Acyltransferase (LCAT). Dieses aus der Leber stammende Enzym verestert das Cholesterin aus peripheren Geweben, aus Chylomikronen und VLDL-Restkörpern mit dem Lecithin (Phosphatidylcholin) in den HDL. So kann mehr Cholesterin in die HDL eingelagert werden. Aktiviert wird die LCAT von Apolipoprotein A1.
Freie Fettsäuren werden nicht von Lipoproteinen transportiert. Sie lagern sich besonders an Albumin an, das mit 60 Gew.% die quantitativ wichtigste Fraktion der Plasmaproteine darstellt (3,5 – 4,5 g/dl Plasma). Albumin kann sieben Fettsäuren gleichzeitig binden und dient auch verschiedenen anderen lipophilen Substanzen als Vehikel. Für Sexualsteroide steht neben dem Albumin auch das Sex hormone binding globulin für den Transport im Körperkreislauf zur Verfügung.
Pathobiochemie
BearbeitenDas Verhältnis von HDL zu LDL beeinflusst das Atheroskleroserisiko (Günstig ist hohes HDL und niedriges LDL). Der Einfluss der Nahrungsaufnahme auf den Cholesterinspiegel ist gegenüber der endogenen Cholesterinbiosynthese jedoch gering.
Biosynthese gesättigter Fettsäuren
BearbeitenAllgemeines
BearbeitenFettsäuren werden synthetisiert als Energiespeicher für „schlechte Zeiten“ (Depotfett) und zur Bildung von Strukturelementen wie Membranlipiden und Baufett.
ATP-abhängige Carboxylierung von Acetyl-CoA zu Malonyl-CoA
Bearbeiten| Tr. | Kov. | All. | ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|---|---|---|
| + Glucose, Insulin - Acyl-CoA |
- AMP-kontrollierte Phosphorylierung | + Citrat - Acyl-CoA |
ATP, CO2
ADP + Pi |
Biotin | Acetyl-CoA- Carboxylase | 6.4.1.2 | Lig | |||
Für die Verlängerung der wachsenden Fettsäurekette wird Malonyl-CoA benötigt. Dieses wird Biotin-abhängig und unter ATP-Verbrauch durch Carboxylierung aus Acetyl-CoA gebildet. Die Reaktion ist das Tor zur Fettsäurebiosynthese, das Schrittmacherenzym Acetyl-CoA-Carboxylase wird dementsprechend transkriptionell, kovalent und allosterisch reguliert.
Beladung des Multienzymkomplexes (MEC) mit einem Starter-Acetyl-Rest
Bearbeiten| Tr. | ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|---|
|
||||||||
| + Glucose, Insulin - cAMP, LCFA |
Acetyl-CoA | Acetyl-CoA | ACP-S-Acetyltransferase | 2.3.1.38 | Tr | |||
| Fettsäure-Synthase (MEC) | 2.3.1.85 | |||||||
|
||||||||
Bevor der Synthesezyklus beginnen kann muss der Multienzymkomplex mit einem Acetyl-Rest als Startermolekül für die Kettenverlängerung beladen werden. Diesen Acetyl-Rest findet man in der fertigen Fettsäure dann ganz am Ende, bei der Synthese von Palmitinsäure z.B. in Form der C-Atome 15 und 16.
Der Synthesezyklus
BearbeitenWiederholung des Zyklus:
| Tr | ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||||
| + Glc, Insulin - cAMP, LCFA |
Malonyl-CoA | Malonyl-CoA | ACP-S-Malonyltransferase | 2.3.1.39 | Tr | |||||||||
| Fettsäure-Synthase (MEC) | 2.3.1.85 | |||||||||||||
|
||||||||||||||
| + Glc, Insulin - cAMP, LCFA |
|
β-Ketoacyl-ACP-Synthase I | 2.3.1.41 | Tr | ||||||||||
| Fettsäure-Synthase (MEC) | 2.3.1.85 | |||||||||||||
|
||||||||||||||
| + Glc, Insulin - cAMP, LCFA |
NADPH/H+ | NADPH/H+ | 3-Ketoacyl-ACP-Reduktase | 1.1.1.100 | Ox | |||||||||
| Fettsäure-Synthase (MEC) | 2.3.1.85 | |||||||||||||
|
||||||||||||||
| + Glc, Insulin - cAMP, LCFA |
|
|
3-Hydroxypalmitoyl-ACP-Hydratase | 4.2.1.61 | Ly | |||||||||
| Fettsäure-Synthase (MEC) | 2.3.1.85 | |||||||||||||
|
||||||||||||||
| + Glc, Insulin - cAMP, LCFA |
NADPH/H+ | NADPH/H+ | Enoyl-ACP-Reduktase | 1.3.1.10 | Ox | |||||||||
| Fettsäure-Synthase (MEC) | 2.3.1.85 | |||||||||||||
|
||||||||||||||
Alle Schritte der Fettsäurebiosynthese erfolgen am Multienzymkomplex (MEC). Zuerst wird Malonyl-CoA auf den bereits mit einem Acetyl- oder Acyl-Rest beladenen MEC übertragen. Malonyl-CoA wird dann decarboxyliert und der Ac(et)yl-Rest auf diesen C2-Rest übertragen. In den folgenden 3 Schritten (Reduktion - Dehydratisierung - Reduktion) wird die Ketogruppe NADPH-abhängig am C3-Atom entfernt und der Kettenabschnitt in den lipophilen gesättigten Kohlenwasserstoffrest ebenfalls NADPH-abhängig umgewandelt.
Abspaltung der fertigen gesättigten Fettsäure
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
|
|||||||
| H2O
[ACP] |
Oleoyl-ACP-Hydrolase (Acyl-ACP-Hydrolase) | 3.1.2.14 | Hyd | ||||
Der Zyklus wird bis zur gewünschten Länge der Fettsäure - meist resultiert Palmitinsäure mit 16 C-Atomen - wiederholt und die Fettsäure dann abgespalten.
Abläufe
BearbeitenIm Organismus werden Fettsäuren v.a. im Fettgewebe und in der Leber synthetisiert, sowie in der Mamma lactans. Bezogen auf die einzelne Zelle findet die Biosynthese gesättigter Fettsäuren bei Eukaryonten an einem Multienzymkomplex (MEC) im Zytosol statt. Der MEC besitzt ein Acyl-Carrier-Protein [ACP] mit zwei wichtigen funktionellen Sulfid-Gruppen, einer peripheren SH-Gruppe (SHp), die von einem Cysteinylrest gebildet wird, und einer zentralen SH-Gruppe (SHz), die von der prosthetischen Gruppe 4'-Phosphopantethein (Phosphat-Pantothenat-Cysteamin) stammt.
Eine reine Umkehrung der β-Oxidation (mit Umkehr der β-Thiolase-Reaktion) ist aus energetischen Gründen nicht möglich. Die Umkehrung gelingt erst durch die Integration einer Decarboxylierung in den Reaktionszyklus. Dafür wird zur Kettenverlängerung nicht Acetyl-CoA, sondern Malonyl-CoA verwendet. Die aus 3 C-Atomen bestehende Dicarbonsäure wird am [ACP] decarboxyliert und reagiert dann als Carbanion leicht mit dem Carbonylkohlenstoff (C=0) der Acyl-Gruppe. Dadurch verschiebt sich das Reaktionsgleichgewicht hin zur Kondensation. Außerdem wird für die Reduktion NADPH/H+ statt NADH/H+benutzt.
Als Startermolekül der Acylbiosynthese dient ein Acetyl-Rest, mit dem der MEC-Komplex beladen wird. Nach Durchlaufen des ersten Zyklus wiederholt sich die Reaktion mit dem nun bereits beladenen Acyl-[ACP]. Mit jedem Zyklus wird die Kette um 2 C-Atome verlängert.
Durch die Verwendung von Propionyl-CoA als Startermolekül entstehen ungeradzahlige Fettsäuren.
Die Reaktionen am bereits beladenen [ACP]-MEC sind wie folgt:
- Malonyltransfer auf den Enzymkomplex
- Kondensation: Übertragung des Acylrests (Cn) der peripheren SH-Gruppe auf den Malonylrest (C3) der zentralen SH-Gruppe unter Abspaltung von CO2 (-C1). Dadurch entsteht eine Ketoacylgruppe (Cn+2).
- Reduktion zur D-Hydroxyacylgruppe.
- Dehydratisierung zum Enoyl.
- Nochmalige Reduktion zum Fettsäurerest.
Der Acyl-Rest wird auf die periphere SH-Gruppe übertragen und der Zyklus beginnt von vorne. Ist die gesättigte Fettsäure lang genug (bis zu 16 C-Atome (Palimitinsäure)), so wird sie durch die Acyl-ACP-Hydrolase vom [ACP] abgespalten.
Regulation
Bearbeiten- Pyruvatdehydrogenase-Reaktion: Die Aktivität der PDH reguliert das Angebot an Acetyl-CoA für Citratzyklus und Fettsäurebiosynthese. Die PDH im Adipozyten wird insbesondere durch Insulin aktiviert (Dephosphorylierung des PDH-Komplexes). Das Peptidhormon steigert zusätzlich den Substratzufluss durch Translokation von GLUT4 in die Zellmembran, so dass Glucose vermehrt in die Zelle diffundieren kann.
- Der geschwindigkeitsbestimmende Schritt der eigentlichen Fettsäurebiosynthese ist die irreversible Acetyl-CoA-Carboxylase-Reaktion, die allosterisch und kovalent reguliert wird. Bei ausreichendem Acyl-CoA-Angebot oder Energiemangel (AMP) wird das Enzym gebremst. Auf der Ebene der Gentranskription wird das Enzym durch Glucose und Insulin induziert und durch Acyl-CoA reprimiert.
- Die Fettsäure-Synthase wird über ihre Transkription reguliert. Insulin und Glucose induzieren die Genexpression über den Transkriptionsfaktor SREBP. Katecholamine und Glucagon hemmen die Expression über cAMP. Langkettige Fettsäuren (LCFA), v.a. mehrfach ungesättigte, hemmen ebenfalls das Ablesen des Gens.
Unterschiede zwischen zytosolischer Fettsäuren-Biosynthese und mitochondrialer β-Oxidation
BearbeitenEin Synthesezyklus enthält wie der β-Oxidations-Zyklus auch zwei Redoxreaktionen (+/- H) und eine Hydratasereaktion (+/- H2O). Die Biosynthese ist auch abhängig von NADPH/H+, während die β-Oxidation kein NADP+ als Redoxpartner benötigt. Diese und weitere Unterschiede in der folgenden Tabelle:
| Zytosolische Fettsäuren-Biosynthese | β-Oxidation | |
|---|---|---|
| Ort | Zytosol | Mitochondrium (Peroxisom) |
| Beteiligte Enzyme | Acetyl-CoA-Carboxylase, Multienzymkomplex | Enzyme, z.T. trifunktionelles Protein |
| Acyl-Carrier | [ACP] (SHp und SHz) | CoA-SH |
| C-Fragmente | Malonyl-CoA, Starter-Acetyl-CoA | Acetyl-CoA |
| Hydroxyl-Intermediate | D | L |
| Redox-Carrier | oxidiert NADPH/H+ | reduziert FAD und NAD+ |
Gewinnung des NADPH/H+ - HMP-Weg und Citrat-Malat-Pyruvat-Zyklus
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
| NADH/H+ | NADH/H+ | Malat-Dehydrogenase
Zytosol |
1.1.1.37 | Ox | |||
| NADP+
CO2, NADPH |
Malat-Enzym
Zytosol |
1.1.1.40 | Ox | ||||
| ATP, HCO3-
ADP, Pi |
Biotin; Mn od. Zn | Pyruvat-Carboxylase
Mitochondrium |
6.4.1.1 | Lig | Pyruvat-Carboxylase-Defizienz | ||
Das für die Biosynthese benötigte NADPH/H+ wird zum großen Teil im Pentosephosphatweg generiert, der wie die Fettsäurensynthese im Zytosol lokalisiert ist.
Daneben kann NADPH/H+ auch im hier dargestellten Citrat-Malat-Pyruvat-Zyklus (Ball-Zyklus) aus NADH/H+ (z.B. aus der Glycolyse) gebildet werden. Das kostet zwei ATP pro NADPH/H+ („bezahlt“ wird an der Pyruvat-Carboxylase und an der ATP-Citrat-Synthase (s.u.)). Der Zyklus verteilt sich auf das zytosolische und das mitochondriale Kompartiment. Für Pyruvat gibt es einen Membrantransporter in der inneren Mitochondrienmembran. Oxalacetat wird in Form von Citrat zurück ins Zytosol transportiert (s.u.). Im Einzelnen laufen folgende Reaktionen ab:
- Oxalacetat wird nach Transport ins Zytosol von der Malat-Dehydrogenase zum Malat reduziert.
- Malat wird vom Malat-Enzym zum Pyruvat oxidiert und decarboxyliert. Die Decarboxylierung liefert genug Energie, um damit NADPH/H+ zu erzeugen.
- Pyruvat wird ins Mitochondrium transportiert und dort unter ATP-Verbrauch wieder zu Oxalacetat carboxyliert.
- Oxalacetat wird - wie im nächsten Abschnitt dargestellt - für den Rücktransport von der Citrat-Synthase mit Acetyl-CoA zum Citrat kondensiert, über die Membran geschafft, und im Zytosol wieder von der ATP-Citrat-Synthase unter ATP-Verbrauch thiolytisch gespalten.
Transport von Acetyl-CoA und Oxalacetat ins Zytosol
Bearbeiten| All. | ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||||
| - ATP, NADH, Citrat | H2O | Citrat-Synthase
Mitochondrium |
2.3.3.1 | Tr | ||||||||||
|
||||||||||||||
| CoA-SH, ATP
ADP + Pi |
ATP-Citrat-Synthase
Zytosol |
2.3.3.8 | Tr | |||||||||||
|
||||||||||||||

Für Acetyl-CoA und Oxalacetat gibt es in der inneren Mitochondrienmembran keinen eigenen Transportmechanismus, so dass die Zelle hier auf Alternativen zurückgreifen muss. Eine solche ist die Kondensation von Acetyl-CoA mit Oxalacetat zu Citrat (1. Reaktion des Citratzyklus). Citrat kann nun den Tricarboxylatcarrier nutzen, um ins Zytosol zu gelangen und dort wieder unter ATP-Verbrauch in Oxalacetat und Acetyl-CoA gespalten werden.
Oxalacetat kann für den Rückweg wie oben dargestellt entweder nur zu Malat reduziert oder zu Pyruvat decarboxyliert werden. Malat kann mit dem Tricarboxylatcarrier zurück ins Mitochondrium gelangen und dort wieder zu Oxalacetat oxidiert werden. Pyruvat kann einen Pyruvat-Transporter nutzen und ebenfalls zum Oxalacetat carboxyliert werden (1. Schritt der Gluconeogenese), so dass auch hier der Kreis geschlossen wird.
Zusammenfassung der Beziehungen zwischen Fettsäurebiosynthese und Glucoseabbau
BearbeitenGlycolyse ⇒ Pentose- -> NADPH/H+ -> Fettsäure-
phosphat- biosynthese Zytosol
⇓ ⇐ weg
⇑
Pyruvat ⇐ Malat ⇔ Oxalacetat + Acetyl-CoA
-- ⇓ ------ ⇓ --------------- ⇑ -------------------------------- Innere mitochondriale Membran
PC
/ ⇒ Oxalacetat
Pyruvat + ⇒ Citrat Mitochondriale
\ ⇒ Acetyl-CoA Matrix
PDH
Bei gutem Glucose-Angebot werden nicht nur die Glycogenspeicher in Muskel und Leber aufgefüllt, sondern durch das vermehrt anfallende Acetyl-CoA auch die Fettreserven. Die Glycolyse findet im Zytosol statt, die dehydrierende Decarboxylierung von Pyruvat durch die Pyruvatdehydrogenase (PDH) jedoch im Mitochondrium. Da die Fettsäuren im Zytosol synthetisiert werden, muss das Acetyl-CoA wieder dorthin geschafft werden. Acetyl-CoA kann die mitochondriale Membran nicht überwinden und wird daher zusammen mit Oxalacetat in Form von Citrat transportiert (Tricarboxylat-Carrier), wie oben dargestellt und in dieser Grafik noch einmal zusammengefasst.
Bei kataboler Stoffwechsellage können diese Transportmechanismen ebenfalls genutzt werden. Nicht im Fettgewebe, sondern in der Leber fließt dann das im Zytosol aus Citrat freigesetzte Oxalacetat in die Gluconeogenese und Acetyl-CoA in die Ketonkörperproduktion.
Weblinks
Bearbeiten- KEGG: Fatty acid biosynthesis - Homo sapiens (human)
- The chemical logic behind... fatty acid metabolism von Prof. Doutor Pedro Silva
- RCSB PDB: Fatty Acid Synthase
Fettsäurenverlängerung in Mitochondrien
BearbeitenAllgemeines
BearbeitenMitochondrien können Fettsäuren z.T. verlängern. Die Reaktionen entsprechen weitgehend einer Umkehrung der β-Oxidation.
Fettsäurenverlängerung
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
Acetyl-CoA |
|
Acetyl-CoA-C-Acyltransferase | 2.3.1.16 | Tr | TFP-Def. | |||||
|
||||||||||
| NADH/H+ | NADH/H+ | 3-Hydroxyacyl-CoA-Dehydrogenase (C6-C10) | 1.1.1.35 | Ox | HADH-Def. | |||||
| Long-chain-3-Hydroxyacyl-CoA-Dehydrogenase | 1.1.1.211 | TFP-Def., LCHAD-Def. | ||||||||
|
||||||||||
|
|
|
Enoyl-CoA-Hydratase | 4.2.1.17 | Ly | TFP-Def. | |||||
|
||||||||||
| NADPH/H+ | NADPH/H+ | Trans-2-Enoyl-CoA-Reduktase (NADPH) | 1.3.1.38 | Ox | ||||||
|
||||||||||
Mitochondrien sind in der Lage Fettsäuren mit mindestens 4 C-Atomen (Butansäure) bis zum Hexadecanoat (Palmitat) zu verlängern. Die Elongation erfolgt dabei mit Acetyl-CoA statt mit Malonyl-CoA. Die Mitochondrien bedienen sich dabei überwiegend der Enzyme der β-Oxidation, es handelt sich also weitgehend um eine rückwärts ablaufende β-Oxidation, die sich von der zytosolischen Fettsäure-Biosynthese in einigen Punkten unterscheidet.
Abspaltung der fertigen gesättigten Fettsäure
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| H2O | Palmitoyl-Protein-Hydrolase | 3.1.2.22 | Hyd | Infantile neuronale Ceroidlipofuscinose 1 (CLN1) | ||||||
|
||||||||||
Weblinks
Bearbeiten
Biosynthese ungesättigter Fettsäuren
BearbeitenAllgemeines
BearbeitenUngesättigte Fettsäuren können z.T. vom Körper gebildet werden, zum großen Teil müssen sie mit der Nahrung aufgenommen werden. Sie beeinflussen als Membranbestandteil deren Fluidität. Die 4fach ungesättigte Fettsäure Arachidonsäure ist Ausgangspunkt für die Biosynthese einer großen Gruppe von Signalmolekülen, den Eikosanoiden.
Biosynthese und Eigenschaften
BearbeitenDas Einfügen von Doppelbindungen in Fettsäuren, die aus Acetyl-CoA synthetisiert oder mit der Nahrung aufgenommen wurden wird gewährleistet durch sog. Desaturasen, die man vorwiegend in der Leber findet, unter Beteiligung von Cytochrom b5. Diese können unter Verbrauch von NADPH/H an den Positionen Δ5, Δ6 und Δ9 Doppelbindungen erzeugen. So kann sich der Körper z.B. die einfach ungesättigten Fettsäuren Palimitoleinsäure (16:1 Δ9) und Ölsäure (18:1 Δ9) synthetisieren. Doppelbindungen über Δ9 hinaus können vom Menschen nicht erzeugt werden, während es Pflanzen an Δ12 und Δ15 noch möglich ist. Die tierische Zelle kann allerdings mit CoA-SH veresterte Fettsäuren am Carboxylende um jeweils 2 C-Atome verlängern oder verkürzen und damit Doppelbindungen verlagern. Viele insbesondere mehrfach ungesättigte Fettsäuren müssen jedoch mit der Nahrung zugeführt werden.
Die ungesättigten Fettsäuren sind meist cis-konfiguriert (cis-Fettsäuren) und die Doppelbindungen sind nicht konjugiert.
Essentielle Fettsäuren:
- ω-3-Fettsäuren
- α-Linolensäure (18:3 Δ9, Δ12, Δ15)
- Eicosapentaensäure (begrenzt aus α-Linolensäure synthetisierbar, 20:5 Δ5, Δ8, Δ11, Δ14, Δ17).
- Docosahexaensäure (begrenzt aus α-Linolensäure synthetisierbar, 22:6 Δ4, Δ7, Δ10, Δ13, Δ16, 19Δ)
- ω-6-Fettsäuren
- Linolsäure (18:2 Δ9, Δ12)
- Octadecatrienoyl-CoA (begrenzt aus Linolsäure synthetisierbar, 18:3 Δ6, Δ9, Δ12)
- Arachidonsäure (begrenzt aus Linolsäure synthetisierbar, 20:4 Δ5, Δ8, Δ11, Δ14)
ω-3-, ω-6- und ω-9-Fettsäuren bilden jeweils eigene Gruppen (bei der Omega-Benennung wird vom Ende her gezählt, so bleibt die Benennung auch nach einer Kettenverlängerung gültig). Innerhalb dieser Gruppen sind Umwandlungen möglich. Die Biosynthese ungesättigter Fettsäuren beginnt bei Pflanze und Tier immer an Position Δ9. Eine weitere Doppelbindung kann der tierische Organismus dann an Position Δ6 einfügen. Um weitere Doppelbindungen zu generieren, muss die Fettsäure zuerst am COOH-Ende um ein C2-Rest verlängert werden. Die Kettenverlängerung am endoplasmatischen Retikulum der Leber erfolgt ähnlich der zytosolischen Fettsäurebiosynthese (NADPH/H+-abh., Malonyl-CoA, jedoch kein MEC), die Kettenverlängerung in Mitochondrien erfolgt analog einer rückwärts ablaufenden β-Oxidation.
Bsp.: Bildung von Arachidonsäure aus Linolsäure
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
|
|||||||
| O2, NADPH/H+
2 H2O, NADP+ |
2Fe+3, Cyto- chrom b5 | Linoleoyl-CoA-Desaturase | 1.14.19.3 | Ox | |||
|
|||||||
| Malonyl-/Acetyl-CoA, 2 NAD(P)H/H+ | Elongationsenzyme (s.o.), Zytosolisch | Tr/Ox/Ly/Ox | |||||
|
|
|||||||
| O2, NADPH/H+
2 H2O, NADP+ |
2Fe+3, Cyt.b5 | Delta(5)-Acyl-CoA-Desaturase | 1.14.19.- | Ox | |||
|
|
|||||||
Weblinks
Bearbeiten- KEGG: Polyunsaturated fatty acid biosynthesis - Homo sapiens (human)
- KEGG: Linoleic acid metabolism - Homo sapiens (human)
Abbau gesättigter Fettsäuren
BearbeitenAllgemeines
BearbeitenFettsäuren werden zur Energiegewinnung vorwiegend in den Mitochondrien in der sog. β-Oxidation oxidiert. Dafür müssen die Fettsäuren zuerst mit Coenzym A aktiviert und mittels Carnitin über die innere Mitochondrienmembran in die Matrix geschafft werden.
Aktivierung der Fettsäuren zu Acyl-CoA im Zytosol
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
| ATP
PPi |
Acyl-CoA-Synthetase | 6.2.1.3 | Lig | Unspezifische X-gebundene mentale Retardierung Typ 63 | |||
|
|
|||||||
| CoA-SH
AMP |
|||||||

Fettsäuren im Zytosol stammen aus der zelleigenen Biosynthese, aus der Lipolyse oder werden aus dem Blut aufgenommen. Damit sie weiterverstoffwechelt werden können müssen sie mit Coenzym A aktiviert werden.
Transport ins Mitochondrium
Bearbeiten| Tr. | All. | ⇓ | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||||||||
| + Lang- kettige FS + T3,T4 |
- Malonyl-CoA | Carnitin-O- Palmitoyltransferase | 2.3.1.21 | Tr | CPT1A-Def., CPT2-Def. | ||||||||||||
|
|||||||||||||||||
| Carnitin-Acylcarnitin- Translocase | CACT-Def. | ||||||||||||||||



Der Abbau von Fettsäuren erfolgt überwiegend im Mitochondrium.
Da langkettige Fettsäuren die innere Mitochondrienmembran nicht überwinden können (die äußere Membran ist sehr durchlässig), wird die Fettsäure von CoA-SH auf Carnitin übertragen und nach Transport über die Membran wieder mit CoA-SH verestert. Die Bildung von Acyl-Carnitin durch die Carnitin-O-Palmitoyltransferase ist der geschwindigkeitsbestimmende Schritt der Fettsäureoxidation.
Die Carnitin-O-Palmitoyltransferase (CPT) wird durch Malonyl-CoA, dem Substrat der Fettsäurenbiosynthese gehemmt. Dadurch wird eine unsinnige gleichzeitige Aktivierung des Fettsäurenauf- und abbaus verhindert.
β-Oxidation
BearbeitenDer Abbau ungesättigter Fettsäuren erfolgt überwiegend in der sog. β-Oxidation in der mitochondrialen Matrix:
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
| FAD | FAD | FAD | (VLC-)Acyl-CoA-Dehydrogenase | 1.3.8.8 | Ox | VLCAD-Def. | |
| (MC-)Acyl-CoA-Dehydrogenase | 1.3.8.7 | MCAD-Def. | |||||
| Acyl-CoA-Oxidase | 1.3.3.6 | Pseudoneonatale Adrenoleukodystrophie | |||||
| (SC-)Butyryl-CoA-Dehydrogenase | 1.3.99.2 | SCAD-Def. | |||||
|
|
|||||||
| H2O
|
H2O
|
Enoyl-CoA-Hydratase | 4.2.1.17 | Ly | TFP-Def. | ||
|
|
|||||||
| NAD+ | NAD+ | NAD | LC-3-Hydroxyacyl-CoA-Dehydrogenase (LCHAD) | 1.1.1.211 | Ox | TFP-Def., LCHAD-Def. | |
| 3-Hydroxyacyl-CoA-Dehydrogenase | 1.1.1.35 | HADH-Def. | |||||
|
|
|||||||
| CoA-SH
|
|
CoA-SH
|
Acetyl-CoA-C-Acyltransferase (β-Ketothiolase) | 2.3.1.16 | Tr | TFP-Def. | |
| Acetyl-CoA-C-Acetyltransferase | 2.3.1.9 | alpha-Methylacetoaceturie | |||||
Der Abbau ungesättigter Fettsäuren erfolgt in der sog. β-Oxidation in der mitochondrialen Matrix. Die letzten drei Schritte werden z.T. von einem einzigen trifunktionellen Protein (TFP) katalysiert.
Die β-Oxidation umfasst vier Schritte. Zuerst wird das Acyl-CoA dehydriert (oxidiert), dann hydratisiert (Wasseranlagerung), noch ein weiteres mal dehydriert und zuletzt ein Acetyl-coA abgespalten. D.h. in einem Zyklus werden zwei Reduktionsäquivalente gewonnen (zwei Oxidationen) und die mit CoA veresterte Fettsäure wird um einen C2-Rest (Acetyl-CoA) gekürzt.
Geradzahlige Fettsäuren werden komplett zu Acetyl-CoA oxidiert. Bei ungeradzahligen Fettsäuren bleibt am Ende ein C3-Rest (Propionyl-CoA) übrig, der gesondert abgebaut wird.
Sehr langkettige Fettsäuren mit mehr als 18 C-Atomen werden zuerst in den Peroxisomen in Teilschritten der β-Oxidation gekürzt, bevor sie ins Mitochondrium verbracht werden. Im Unterschied zur mitochondrialen β-Oxidation werden die Elektronen dort ohne Energiegewinn auf molekularen Sauerstoff übertragen. Das dabei entstehende Wasserstoffperoxid (H2O2) wird durch die Katalase zu Wasser und Sauerstoff disproportioniert.
Energiebilanz
BearbeitenDie Energiebilanz hängt von der Länge der Fettsäure ab. Hier ein Rechenbeispiel für Stearinsäure (C18H36O2).:
| Aktivierung | Stearinsäure -> Stearyl-CoA | - 2 ATP (zwei energiereiche Bindungen, ATP -> AMP + PPi) |
| β-Oxidation | Stearyl-CoA -> 9 Acetyl-CoA | 8 FADH2 + 8 NADH/H+ = 8 x 1,5 ATP + 8 x 2,5 ATP = + 32 ATP |
| Citratzyklus | 9 Acetyl-CoA -> 18 CO2 | 9 x 1 FADH2 + 9 x 3 NADH/H+ + 9 ATP (GTP) = 9 x 1,5 ATP + 9 x 3 x 2,5 ATP + 9 ATP = + 90 ATP |
Aus einer Stearinsäure gewinnt die Zelle also 120 ATP (vergleiche 32 ATP bei Oxidation eines Glucose-Moleküls).(Anm.: Das Ergebnis hängt davon ab, welche Umrechnungsfaktoren man für die Umsetzung der Reduktionsäquivalente in ATP animmt, im o.g. Fall 1,5 für FADH2 und 2,5 für NADH/H+. Manche Lehrbücher rechnen mit den Faktoren 2 für FADH2 und 3 für NADH/H+. Die Bilanz ergäbe dann in unserem Fall 146 ATP.)
Pathobiochemie
BearbeitenGenetische Defekte der beteiligten mitochondrialen Enzyme führen zu einer Verwertungsstörung von Fettsäuren. Diese stehen dann nicht mehr zur Energieversorgung der Zelle zu Verfügung und akkumulieren im Gewebe (Organverfettung). Beides beeinträchtigt die Organfunktion vor allem von Herz, Skelettmuskel, Nervensystem und Leber. Symptome treten meist nach Fastenperioden (Glucosemangel nach Aufbrauchen der Glycogenspeicher) oder in Zeiten erhöhten Energiebedarfs (Infektionen) auf, je nach Schwere des Enzymdefekts häufig schon in der Neonatalperiode oder im frühen Kindesalter. Typischerweise lässt sich dabei eine hypoketotische Hypoglykämie nachweisen.
Defekte können auch Enzyme in Peroxisomen betreffen (Acyl-CoA-Oxidase), die am Abbau sehr langkettiger und/oder ungesättigter Fettsäuren beteiligt sind. Diese manifestieren sich vor allem am Nervensystem.
Der häufigste Defekt des Fettsäurenabbaus ist die MCAD-Defizienz und betrifft damit den ersten Schritt der β-Oxidation.
Eine Besonderheit weist die LCHAD-Defizienz auf: Eine Stoffwechseldefekt des Feten kann hierbei in der Schwangerschaft eine Erkrankung der Mutter in Form einer sog. Gestose (wie z.B. das HELLP-Syndrom oder die akute Leberverfettung in der Schwaangerschaft, kurz AFLP) hervorrufen.
Literatur
BearbeitenDisorders of fatty-acid oxidation (FAOD):
- Lindner M, Hoffmann GF, Matern D. “Newborn screening for disorders of fatty-acid oxidation: experience and recommendations from an expert meeting”. J. Inherit. Metab. Dis., 33:521–6, October 2010. DOI:10.1007/s10545-010-9076-8. PMID 20373143.
- Gregersen N, Andresen BS, Bross P. “Prevalent mutations in fatty acid oxidation disorders: diagnostic considerations”. Eur. J. Pediatr., 159 Suppl 3:S213–8, December 2000. PMID 11216903.
- Rinaldo P, Raymond K, al-Odaib A, Bennett MJ. “Clinical and biochemical features of fatty acid oxidation disorders”. Curr. Opin. Pediatr., 10:615–21, December 1998. PMID 9848022.
Weblinks
Bearbeiten- KEGG: Fatty acid metabolism - Homo sapiens (human)
- The chemical logic behind... fatty acid metabolism von Prof. Doutor Pedro Silva
Abbau von Propionyl-CoA
BearbeitenAus Propionyl-CoA entsteht durch ATP-abhängige Carboxylierung und zweimalige Isomerisierung Succinyl-CoA
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
|
H2O, CO2, ATP ADP + Pi |
Biotin | Propionyl-CoA-Carboxylase | 6.4.1.3 | Lig | Propionacidämie | |||||
|
||||||||||
| Methylmalonyl-CoA-Epimerase | 5.1.99.1 | Iso | Methylmalonyl-CoA-Epimerase-Def. | |||||||
|
||||||||||
| Adenosyl-cobalamin | L-Methylmalonyl-CoA-Mutase | 5.4.99.2 | Iso | Methylmalonylacidurie (MMA), complementation group 'mut' | ||||||
|
||||||||||


Bei der vollständigen β-Oxidation ungeradzahliger Fettsäuren bleibt am Ende Propionyl-CoA übrig. Dieses wird zu Succinyl-CoA carboxyliert und isomerisiert und kann dann leicht im Citratzyklus weiter verstoffwechselt werden. Als Cofaktoren werden Biotin (Vitamin H) für die Carboxylierung und Cobalamin (Vitamin B12) für die Isomerisierung benötigt.
Propionyl-CoA fällt auch beim Abbau der verzweigtkettigen Aminosäuren Isoleucin und Valin und beim Abbau von Methionin und Threonin an sowie im Rahmen der Gallensäuren-Biosynthese.
Weblinks
Bearbeiten
Abbau ungesättigter Fettsäuren
BearbeitenAllgemeines
BearbeitenDie Doppelbindung in ungesättigten Fettsäuren ist cis-konfiguriert, das Intermediat der β-Oxidation jedoch trans (Δ2-trans-Enoyl-CoA). Daher erfordert ihr Abbau die Umlagerung der Doppelbindungen von cis nach trans. Abhängig davon, ob die jeweilige Doppelbindung auf einem geradzahligen oder ungeradzahligen C-Atom liegt, sind hierfür drei oder nur ein Reaktionsschritt erforderlich.
Die Reaktionen im Detail
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
|
|||||||
| FAD+ | Dehydrogenase | ? | Ox | ||||
|
|||||||
| NADPH/H+ | Reduktase | ? | Ox | ||||
|
|||||||
| Δ3-cis-Δ2-trans-Enoyl-CoA-Isomerase | 5.3.3.8 | Iso | |||||
|
|||||||
Ungesättigte Fettsäuren werden bis zur ersten Doppelbindung wie gewohnt in der β-Oxidation abgebaut. Der Zyklus läuft bis ein Δ3-cis-Enoyl-CoA (Fettsäure mit Δungeradzahlig) bzw. ein Δ4-cis-Enoyl-CoA (Fettsäure mit Δgeradzahlig) vorliegt. Da die Doppelbindung in ungesättigten Fettsäuren cis-konfiguriert ist, das Intermediat der β-Oxidation jedoch trans (Δ2-trans-Enoyl-CoA), muss nun die cis-Konfiguration in trans umgewandelt werden.
- Δ3-cis-Enoyl-CoA wird dazu einfach von einer Δ3-cis-Δ2-trans-Enoyl-CoA-Isomerase zum Δ2-trans-Enoyl-CoA isomerisiert.
- Δ4-cis-Enoyl-CoA wird zuerst zum Δ2trans-Δ4-cis-Dienoyl-CoA oxidiert und dann unter Verbrauch von NADPH/H+ zum Δ3-cis-Enoyl-CoA reduziert. Dieses wird dann wie gehabt zum Δ2-trans-Enoyl-CoA isomerisiert.
Die β-Oxidation läuft nun bis zur nächsten Doppelbindung weiter.
Weblinks
Bearbeiten- The chemical logic behind... fatty acid metabolism von Prof. Doutor Pedro Silva
Abbau verzweigtkettiger Fettsäuren
Bearbeiten
Die verzweigtkettige Phytansäure wird vom Körper nicht selbst gebildet, sie ist pflanzlicher Herkunft und wird ausschließlich über die Nahrung aufgenommen z.B. als Bestandteil von Chlorophyll. Da die Methylgruppe am C3-Atom (β-Position) die β-Oxidation unmöglich macht, wird die Fettsäure zuerst in der peroxisomalen α-Oxidation unter Verwendung von molekularem Sauerstoff decarboxyliert. Das Enzym Phytanoyl–CoA-Hydroxylase (EC 1.14.11.18) katalysiert die Reaktion. Als Cofaktoren sind Eisen und Ascorbat beteiligt. Dadurch rutscht die Methylgruppe in die α-Position und die β-Position wird frei. Das entstandene Pristanal kann dann nach Transport ins Mitochondrium auf die herkömmliche Art oxidiert werden.
Pathobiochemie: Beim Morbus Refsum (OMIM) ist entweder die Phytanoyl–CoA-Hydroxylase defizient oder das Protein Peroxin-7, ein Transportprotein, das die Phytanol-CoA-Hydroxylase in das Peroxisom transportiert. Klinische Zeichen umfassen Retinitis pigmentosa, periphere Polyneuropathie, Kleinhirnstörungen, Taubheit, EKG-Veränderungen, Ichthyosis und multiple epiphyseale Dysplasie.
Literatur:
- Wanders RJ, van Grunsven EG, Jansen GA. “Lipid metabolism in peroxisomes: enzymology, functions and dysfunctions of the fatty acid alpha- and beta-oxidation systems in humans”. Biochem. Soc. Trans., 28:141–9, February 2000. PMID 10816116.
- Schofield CJ, McDonough MA. “Structural and mechanistic studies on the peroxisomal oxygenase phytanoyl-CoA 2-hydroxylase (PhyH)”. Biochem. Soc. Trans., 35:870–5, November 2007. DOI:10.1042/BST0350870. PMID 17956235.
Weblinks:
Arachidonsäure-Stoffwechsel
BearbeitenAllgemeines
BearbeitenDie 4fach ungesättigte Fettsäure Arachidonsäure (5,8,11,14-Eicosatetraensäure) ist der Ausgangspunkt für die Biosynthese der sog. Eikosanoide, zu denen die Prostaglandine und Leukotriene gehören.
Prostaglandinbiosynthese
BearbeitenBiosynthese von Prostaglandin G2 und H2
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | ||
|---|---|---|---|---|---|---|---|---|---|
|
|||||||||
| H2O
Monoacylphosphoglycerin |
Ca | Phospholipase A2 | 3.1.1.4 | Hyd | |||||
|
|||||||||
| 2 O2
|
Häm | Prostaglandin-Endoperoxid-Synthase (Cyclooxygenase-Aktivität) | 1.14.99.1 | Ox | |||||
|
|||||||||
| AH2
H2O, A |
Häm | Prostaglandin-Endoperoxid-Synthase (Peroxidase-Aktivität) | 1.14.99.1 | Ox | |||||
|
|||||||||




Aus PGH2 werden die anderen Prostaglandine gebildet. Durch Isomerisierung entsteht TxA2, PGI2 (Prostacyclin), PGD2 oder PGE2. Die Synthese von PGF2α und 11-epi-PGF2α erfordert zusätzlich noch eine Reduktion.
Biosynthese von Thromboxan A2
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
|
|||||||
| Häm-Thiolat | Thromboxan-A-Synthase (CYP5A1) | 5.3.99.5 | Iso | Ghosal haemato-diaphyseal dysplasia | |||
|
|||||||

Biologische Wirkung: Thrombozytenaggregation, Vasokonstriktion. TxA2 wird v.a. von Thrombozyten gebildet (COX1).
Biosynthese von Prostacyclin (Prostaglandin I2)
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
|
|||||||
| Häm-Thiolat | Prostacyclin-Synthase | 5.3.99.4 | Iso | ||||
|
|||||||

Biologische Wirkung: Vasodilatation, Hemmung der Plättchenaggregation. Prostacyclin ist der Gegenspieler des TxA2 und wird vom Endothel gebildet (COX2).
Biosynthese von Prostaglandin D2
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
|
|||||||
| Glutathion | PGD-Synthase | 5.3.99.2 | Iso | ||||
|
|||||||
| NADPH/H+ | PGF-Synthase | 1.1.1.188 | Ox | ||||
|
|||||||


Biologische Wirkung: PGD2 fungiert im ZNS als Neuromodulator und trophischer Faktor, beeinflusst den Tonus der glatten Muskulatur und hemmt die Plättchenaggregation. Weiterhin ist es evtl. in den Ablauf des Non-REM-Schlafs involviert.
Biosynthese von Prostaglandin E2 und F2α
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
|
|||||||
| Glutathion | PGE-Synthase | 5.3.99.3 | Iso | ||||
|
|||||||
| NADPH/H+ | Carbonyl-Reduktase (NADPH) | 1.1.1.184 | Ox | ||||
| PGE2- 9-Reduktase | 1.1.1.189 | ||||||
|
|||||||


Biologische Wirkung:
- PGE2 wird durch den Entzündungsmediator Interleukin-1β (IL1β) und das Tumorsuppressorprotein TP53 induziert. Es ist u.a. beteiligt am Entzündungsschmerz. PGE2 ist weiterhin für die Erzeugung von Fieber verantwortlich: Bakterienbestandteile und IL-1 stimulieren das Epithel des Organum vasculosum laminae terminalis (OVLT) -> Expression von COX-2↑ -> PGE2-Synthese↑ -> Stimulation des Temperaturregulationszentrums im Nucleus preopticus medianus des Hypothalamus über EP3-Rezeptoren -> Erhöhung des Temperatur-Sollwertes und Aktivierung der Wärmeproduktion (Kältegefühl, Muskelzittern, erhöhte Stoffwechselaktivität). Im Magen wirkt PGE2 schleimhautprotektiv und hemmt die Bildung der Magensäure. Am Uterus führt es zu Kontraktionen.
- PGF2α wirkt luteolytisch und ist an der Regulation des Augeninnendrucks beteiligt und an der Uteruskontraktion.
Leukotrienbiosynthese
BearbeitenBiosynthese der Leukotriene A4 und B4
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| H2O
Monoacylphosphoglycerin |
Ca | Phospholipase A2 | 3.1.1.4 | Hyd | ||||||
|
||||||||||
| O2
|
Fe | Arachidonat-5-Lipoxygenase | 1.13.11.34 | Ox | ||||||
|
||||||||||
|
|
Fe | Arachidonat-5-Lipoxygenase | 1.13.11.34 | Ox | ||||||
|
||||||||||
| H2O
|
Zn | LTA4-Hydrolase | 3.3.2.6 | Hyd | ||||||
|
||||||||||



Biologische Wirkung: LTB4-Rezeptoren finden sich v.a. im lymphatischen Gewebe und weisen auf eine immunmodulatorische Rolle hin.
Biosynthese der Leukotriene C4, D4 und E4 aus Leukotrien A4
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
|
|||||||
| Glutathion
|
LTC4-Synthase | 4.4.1.20 | Ly | LTC4-Synthase-Def. | |||
|
|||||||
| Aminosäure
γ-Glutamyl-Aminosäure |
γ-Glutamyltransferase | 2.3.2.2 | Tr | Glutathionurie | |||
|
|||||||
| H2O | Dipeptidase 2 | 3.4.13.19 | Hy | ||||
|
|||||||



 |
Biologische Wirkung: Leukotriene wirken stark proinflammatorisch. Die Leukotriene C4, D4 und E4 wirken konstriktorisch an der glatten Muskulatur, z.B. an der Bronchialmuskulatur und an den Hautgefäßen.
Allgemeines zu den Eikosanoiden
BearbeitenProstaglandine und Leukotriene sind wichtige Mediatoren im lokalen Gewebsstoffwechsel und vermitteln u.a. Entzündungsvorgänge. Sie werden aus der 4fach ungesättigten Fettsäure Arachidonsäure (5,8,11,14-Eicosatetraensäure) gebildet, die durch die Phospholipase A2 von Phospholipiden der Zellmembran, z.B. dem Phosphatidylcholin (Lecithin), abgespalten wird. Arachidonsäure ist essentiell bzw. kann begrenzt aus der essentiellen ω-6-Fettsäure Linolsäure gebildet werden.
Pharmakologie
BearbeitenMit Cyclooxygenase-Hemmern wie Acetylsalicylsäure (ASS) lässt sich die Prostaglandinsynthese unterbinden, was die schmerzlindernde, entzündungshemmende und fiebersenkende Wirkung erklärt. ASS hemmt v.a. das Isoenzym COX1 in den Thrombozyten durch irreversible Acetylierung, die das thrombogene TxA2 bildet. Die Wirkung auf die COX2, die im Endothel v.a. das antithrombogene Prostacyclin (PGI2) produziert, ist geringer. Zudem kann das Endothel das Enzym rasch regenerieren, was den kernlosen Thrombozyten nicht möglich ist. Das Gleichgewicht zwischen TxA2 und PGI2 wird dadurch zum antithrombogenen PGI2 hin verschoben und prädestiniert ASS als Medikament zur Prophylaxe von akuten Gefässverschlüssen, die durch Aufbrechen und Thrombosierung von Atheroskleroseplaques entstehen. Eine häufige Anwendung ist z.B. die Rezidivprophylaxe nach einem Herzinfarkt. Kortikosteroide wie Cortison hemmen die Kaskade an einem früheren Punkt (Hemmung der Phospholipase A2 durch Lipocortin, das von Kortikoiden induziert wird) und unterbrechen damit sowohl die Prostaglandin- als auch die Leukotrien-Biosynthese. Dies - und die Wirkung an vielen anderen Schalthebeln im Organismus - erklärt ihren stärkeren antientzündlichen und immunsuppressiven Effekt. Der Leukotrien D4-Rezeptor-Antagonist Montelukast wird bei Asthma bronchiale angewendet. Das Prostazyklin-Analogon Iloprost findet bei der pulmonalen Hypertonie eine Anwendung. Das vasoaktive Prostaglandin E1 (Alprostadil, Prostavasin) ist parenteral bei schwerer AVK und bei erektiler Dysfunktion verwendbar. Das Prostaglandin E1-Analogon Gemeprost wird vaginal angewendet zur Zervixerweiterung und Förderung der Uteruskontration. Ein weiteres PGE1-Analogon ist das Misoprostol, das früher als Magenschleimhautschutz eingesetzt wurde.
Toxikologie
BearbeitenDie Phospholipase A2 ist ein wirksamer Bestandteil verschiedener Tiergifte und z.B. - neben weiteren Toxinen - im Gift der indischen Kobra (Naja naja) enthalten.
Weblinks
BearbeitenTriacylglycerinbiosynthese
BearbeitenAllgemeines
BearbeitenTriacylglycerine (Triglyceride, Neutralfette) bilden die Hauptmasse des Fettgewebes. Sie werden in Vakuolen in den Fettzellen eingelagert. Fettgewebe hat sowohl eine strukturelle Funktion (Baufett: Wärmeisolation, Stabilisation der Organlage, Lückenfüller) als auch eine Speicherfunktion (Depotfett). Fette enthalten pro Masse doppelt soviel Energie wie Kohlenhydrate oder Eiweiße. Nach dem Glykogen bilden sie die wichtigeste Energiereserve im Nüchtern- oder Hungerstoffwechsel.
Triglyceride entstehen durch Veresterung von Glycerin mit drei Fettsäuren. Hierfür muss Glycerin als Glycerin-3-phosphat vorliegen.
Die Zellen der Darmmukosa bilden Triglyceride aus den resorbierten Fettsäuren, Monoacylglycerinen und Glycerin-Molekülen und bauen diese zusammen mit dem Apolipoprotein B48 in die Chylomikronen ein, die dann über den intestinalen Lymphstrom (Umgehung der Leber) abtransportiert werden.
Erzeugung von Glycerin-3-phosphat (α-Glycerophosphat)
BearbeitenGlycerin-3-phosphat (α-Glycerophosphat) wird erzeugt durch Reduktion von Dihydroxyacetonphosphat (Verbindung der Triacylglycerinsynthese mit der Glycolyse). Alternativ können manche Gewebe (Leber, Niere, Darmmukosa, laktierende Mamma) α-Glycerophosphat durch Phosphorylierung von Glycerin gewinnen.
| ⇓ | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| Triose-phosphat-Isomerase | 5.3.1.1 | Iso | TPI1-Def. (Hämolyt. Anämie, progred. neuro-musk. S.) | |||||||
|
||||||||||
| NADH/H+ | NADH/H+ | Glycerin-3-phosphat-Dehydrogenase | 1.1.1.8 | Ox | ||||||
|
||||||||||
| ADP
ATP |
Glycerol-Kinase | 2.7.1.30 | Tr | Hypergycerinämie | ||||||
|
||||||||||

Bildung der Triacylglycerine
BearbeitenTriacylglycerin (Triglyceride) entsteht aus α-Glycerophosphat durch Veresterung mit 3 (mit CoA-SH aktivierten) Fettsäuren.
| ⇓ | Subs. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| Acyl-CoA | Glycerol-3-phosphat- O-Acyltransferase | 2.3.1.15 | Tr | |||||||
|
||||||||||
| Acyl-CoA | 1-Acylglycerol-3-phosphat- O-Acyltransferase | 2.3.1.51 | Tr | |||||||
|
||||||||||
|
|
Phosphatidat-Phosphatase | 3.1.3.4 | Hyd | |||||||
|
||||||||||
| Acyl-CoA | Diacylglycerin-O-Acyltransferase | 2.3.1.20 | Tr | |||||||
|
||||||||||




Regulation
BearbeitenDie Bildung von Fett ist abhängig von der Nahrungszufuhr und wird durch Insulin stimuliert. Die Fettsäuren können dabei aus der Nahrung stammen oder aus Acetyl-CoA gebildet werden, das seinerseits beim Abbau von Glucose und Aminosäuren entsteht.
Weblinks
Bearbeiten
Triacylglycerinabbau
BearbeitenAllgemeines
BearbeitenDer Abbau von Körperfett (Lipolyse) zu Glycerin und Fettsäuren wird besonders dann stimuliert, wenn die Glucosereserven (Glykogen) erschöpft sind. Im Blut transportierte Acylglycerine müssen ebenfalls gespalten werden damit sie in die Zellen aufgenommen werden können. Selbiges gilt für Nahrungsfette im Magen-Darm-Trakt.
Triacylglycerin wird zu Glycerin und 3 Fettsäuren hydrolytisch gespalten
Bearbeiten| Tr. | Kov. | ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||
| + Phosph.
- Dephos. |
H2O | Hormonsensitive Lipase (HSL) Fettgewebe, Skelett- und Herzmuskel, Pankreas-β-Zellen, Nebenniere, Gonaden | 3.1.1.79 | Hyd | ||||||||
| + Insulin | Lipoprotein-Lipase Endothel | 3.1.1.34 | Hyperlipoproteinämie Typ IA | |||||||||
| Triacylglycerin-Lipase GIT | 3.1.1.3 | Pankreas-Lipase-Def., Hepatische Lipase-Def. | ||||||||||
|
||||||||||||
| + Phosph.
- Dephos. |
H2O | Hormonsensitive Lipase | 3.1.1.79 | Hyd | ||||||||
| Triacylglycerin-Lipase GIT | 3.1.1.3 | Pankreas-Lipase-Def., Hepatische Lipase-Def. | ||||||||||
|
||||||||||||
| + Phosph.
- Dephos. |
H2O | Hormonsensitive Lipase | 3.1.1.79 | Hyd | ||||||||
| Acylglycerin-Lipase | 3.1.1.23 | |||||||||||
|
||||||||||||


Die Hormonsensitive Lipase
BearbeitenBei der Freisetzung von Fettsäuren und Glycerin aus dem Fettgewebe spielt die hormonsensitive Lipase (HSL) die entscheidende Rolle. Das Enzym wird von der Proteinkinase A (Katecholamine, Glucagon -> cAMP↑) phosphoryliert und damit aktiviert. Der Gegenspieler Insulin aktiviert die cAMP-Phosphodiesterase, der cAMP-Spiegel fällt, die Proteinkinase A wird dephosphoryliert und der Fettabbau gebremst.
Eine Proteinphosphatase macht die Vorgänge bei niedrigen cAMP-Spiegeln rückgängig.
Die Lipoprotein-Lipase
BearbeitenDie Lipoprotein-Lipase wird von den Endothelzellen der Blutgefäße exprimiert. Sie spaltet die Triglyceride der fettreichen Chylomikronen, die über den Ductus thoracicus aus dem Verdauungstrakt ins Blut gelangen. Die höchste Dichte haben sie auf den Kapillarenendothelien von Muskel- und Fettgewebe. Ihre Expression wird von Insulin induziert.
Die Pankreas-Lipase
BearbeitenTriglyceride werden im Darm v.a. durch die Pankreas-Lipase in Monoacylglycerine und Fettsäuren gespalten.
Klinische Chemie und Laboratoriumsmedizin
BearbeitenEine Erhöhung der Pankreas-Lipase im Serum kann auf eine Pankreatitis hinweisen. Die häufigsten Ursachen einer Bauchspeicheldrüsenentzündung sind Alkohol und Gallensteine, die den Ausführungsgang bzw. die Papilla Vateri verlegen.
Weblinks
Bearbeiten- KEGG: Glycerolipid metabolism - Homo sapiens (human)
- The chemical logic behind... fatty acid metabolism von Prof. Doutor Pedro Silva
Phosphoglycerid-Stoffwechsel
BearbeitenAllgemeines
BearbeitenPhospholipide bilden den Hauptteil der Lipidphase biologischer (Doppel-)Membranen. Sie bestehen aus einer hydrophilen Phosphatgruppe und meist zwei hydrophoben Fettsäuren, die über ein Glycerin verbunden sind. Man unterscheidet zwei Gruppen, Phosphoglyceride und Sphingomyeline (die Sphingomyeline gehören sowohl zu den Phospholipiden als auch zu den Sphingolipiden, denen ein eigenes Kapitel gewidmet ist).
Wichtige Phosphoglyceride, die am Aufbau von Biomembranen beteiligt sind, sind Phosphatidylethanolamin, Phosphatidylserin, sowie das mengenmäßig am häufigsten vorkommende Phosphoglycerid, das Phosphatidylcholin (Lecithin) und die Phosphatidyl-Inositole, die eine wichtige Rolle in intrazellulären Signaltransduktionskaskaden spielen.
Biosynthese von Phosphatidat (1,2-Diacylglycerin-3-phosphat)
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| NADH/H+ | NADH/H+ | Glycerin-3-phosphat-Dehydrogenase 1 löslich | 1.1.1.8 | Ox | ||||||
|
Ox. Akz. |
oder
|
Ox. Akz. |
Flavin | oder
Glycerol-3-phosphat-Dehydrogenase 2 mitochondrial |
1.1.99.5 | |||||
|
||||||||||
| Acyl-CoA | Glycerol-3-phosphat-Acyltransferase mitochondrial | 2.3.1.15 | Tr | |||||||
|
||||||||||
| Acyl-CoA | 1-Acylglycerol-3-phosphat- O-Acyltransferase | 2.3.1.51 | Tr | |||||||
|
||||||||||
Die aufgeführten Schritte zur Biosynthese von 1,2-Diacylglycerin-3-phosphat (Phosphatidsäure bzw. Phosphatidat) sind mit den ersten Schritten der Triacylglycerinbiosynthese identisch. Phosphatidat kann auch durch Phosphorylierung von 1,2-Diacylglycerin (aus dem Triacylglycerin-Abbau) gewonnen werden (Diacylglycerinkinase, 2.7.1.107).
Phosphatidat ist der Ausgangspunkt für die Biosynthese der verschiedenen Phosphoglyceride. Phosphadidat selbst findet sich im Gewebe nur in geringen Mengen.
Biosynthese von 1-Phosphatidyl-D-myo-Inositol
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| CTP
PPi |
CDP-Diacylglycerol-Synthase (Phosphatidat-Cytidylyltransferase) | 2.7.7.41 | Tr | |||||||
|
||||||||||
| myo-Inositol | CDP-Diacylglycerol--Inositol-3-Phosphatidyltransferase | 2.7.8.11 | Tr | |||||||
|
||||||||||

Den weiteren Stoffwechsel der Inositolphosphate finden Sie im Kapitel Inositolphosphat-Stoffwechsel. 1-Phosphatidyl-D-myo-Inositol ist auch der Startpunkt der Biosynthese von Glycosylphosphatidylinositol-Ankern (GPI-Anker).
Biosynthese von Cardiolipin
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| CTP
PPi |
CDP-Diacylglycerol-Synthase (Phosphatidat-Cytidylyltransferase) | 2.7.7.41 | Tr | |||||||
|
||||||||||
| Glycerin-3-phosphat | CDP-Diacylglycerol--Glycerol- 3-phosphat-3-Phosphatidyltransferase | 2.7.8.5 | Tr | |||||||
|
||||||||||
| H2O
Pi |
Phosphatidylglycerophosphatase | 3.1.3.27 | Hyd | |||||||
|
||||||||||
| CDP-Diacylglycerin | Cardiolipin-Synthase 1 | ? | ||||||||
|
||||||||||



Cardiolipin (Diphosphatidylglycerin) findet sich in hoher Konzentration in der inneren Membran von Mitochondrien und damit auch vermehrt in Mitochondrien-reichen Geweben wie Herz und Muskel.
Klinik: Bei der Syphilis (und vielen anderen Erkrankungen, die mit Gewebsschäden einhergehen) treten Autoantikörper gegen das mitochondriale Cardiolipin (Cl) auf. Diese können mit dem sog. VDRL-Test (Veneral-Disease-Research-Laboratories-Test) in einer Agglutinationsreaktion nachgewiesen und zur Verlaufskontrolle der Syphillis genutzt werden.
Eine erbliche Erkrankung, die mit dem Cardiolipin-Stoffwechsel assoziiert ist das Barth-Syndrom.
Stoffwechsel von Phosphatidylcholin (Lecithin)
BearbeitenAusgangspunkt ist 1,2-Diacylglycerin. Dieses stammt aus dem Triacylglycerin-Abbau oder aus Phosphatidat (1,2-Diacylglycerin-3-phosphat) (s.o. und Triacylglycerinbiosynthese).
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| H2O
Pi |
Phosphatidat-Phosphatase | 3.1.3.4 | Hyd | |||||||
|
||||||||||
| CDP-Cholin | Diacylglycerol-Cholinphosphotransferase | 2.7.8.2 | Tr | |||||||
|
||||||||||
| Sterol
Sterol-Fettsäureester |
Lecithin--Cholesterol-Acyltransferase (LCAT) | 2.3.1.43 | Tr | Fischaugenkrankheit, Norum-Krankheit | ||||||
|
|
oder
|
Ca | oder
Phospholipase A2 |
3.1.1.4 | Hyd | |||||
|
||||||||||
| H2O | Lysophospholipase | 3.1.1.5 | Hyd | |||||||
|
||||||||||



Spaltung von Phosphatidylcholin zu Cholin und Phosphatidat:
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| H2O | Phospholipase D | 3.1.4.4 | Hyd | |||||||
|
||||||||||
Stoffwechsel von Phosphatidylserin und Phosphatidylethanolamin
BearbeitenBiosynthese von Phosphatidylserin und Phosphatidylethanolamin aus Lecithin und sukzessive Abspaltung der Fettsäuren:
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| L-Serin | Phosphatidylserin-Synthase | 2.7.8.- | Tr | |||||||
|
||||||||||
|
|
1. |
Ethanolamin | Pyridoxal- phosphat od. Pyruvat | 1) Phosphatidylserin- Decarboxylase | 4.1.1.65 | Ly | ||||
| 2) Phosphatidylserin- Synthase 2 | 2.7.8.- | Tr | ||||||||
|
||||||||||
| H2O | Ca | Phospholipase A2 | 3.1.1.4 | Hyd | ||||||
|
||||||||||
| H2O | Lysophospholipase | 3.1.1.5 | Hyd | |||||||
|
||||||||||




Spaltung von Phosphatidylethanolamin zu Ethanolamin und Phosphatidat:
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| H2O
Ethanolamin |
Phospholipase D | 3.1.4.4 | Hyd | |||||||
|
||||||||||
Phosphatidylethanolamin und Phosphatidylcholin (Lecithin) besitzen eine negativ geladene Phosphat-Gruppe (Anion) und ein positiv geladenes Stickstoff-Atom (Kation), d.h. sie sind insgesamt neutral geladen. Phosphatidylserin trägt zusätzlich eine negative geladene Carboxyl-Gruppe und ist deswegen im physiologischen Milieu negativ geladen.
Weblinks
Bearbeiten
Cholin-Stoffwechsel
BearbeitenAllgemeines
BearbeitenCholin entspricht chemisch einem Ethanolamin, das am Stickstoffatom 3fach methyliert wurde.
Bildung und Abbau von Acetylcholin
Bearbeiten| ⇓ | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
|
|||||||
| Acetyl-CoA | 1. |
H2O |
1) Cholin-O-Acetyltransferase | 2.3.1.6 | Tr | Kongenitales Myasthenie-Syndrom mit episodischer Apnoe (CMS-EA) | |
| 2) Acetylcholinesterase | 3.1.1.7 | Hyd | |||||
|
|||||||


 |
 |
Das kationische Acetylcholin ist ein wichtiger Neurotransmitter an der Muskelendplatte, im Gehirn, in den vegetativen Ganglien, an den parasympathischen Nervenenden sowie an der sympathischen Schweißdrüseninnervation. Im synaptischen Spalt wird es von der Acetylcholinesterase inaktiviert.
Pharmakologie und Toxikologie: Die Acetylcholinesterase kann durch Medikamente (Acetylcholinesterasehemmer, indirekte Parasympathomimetika) gehemmt werden. Damit können z.B. die postoperative Darmatonie (durch cholinerge Anregung der Darmmotilität), das Glaukom (durch Verengung der Pupille mit Erweiterung des Kammerwinkels), die Myasthenia gravis (durch Erhöhung der Acetylcholinkonzentration an der Muskelendplatte) und die Alzheimerdemenz (durch Erhöhung des kortikalen Acetylcholinangebots) behandelt werden. Weiterhin kann damit die Wirkung von Atropin (ein Muskarinrezeptorblocker) und nicht-depolarisierenden Muskelrelaxantien (Acytylcholinrezeptorblocker) antagonisiert werden. Zu den Giften, die hier angreifen, gehören z.B. die Alkylphosphate, die die Acetylcholinesterase durch Phosphorylierung längerfristig hemmen. Zu dieser Gruppe gehören das Parathion (E605), früher als Insektenvertilger eingesetzt, und die Kampfgase Tabun, Sarin und VX. Die Vergiftung führt zum cholinergen Syndrom mit Muskelkrämpfen und vegetativer Entgleisung (Miosis, Schwitzen, Hypersalivation, Bronchialhypersekretion und -spasmen, Bradykardie, Blutdruckabfall, Diarrhoe und Bauchkrämpfe, Harndrang).
Biosynthese von Phosphocholin und CDP-Cholin
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
|
|||||||
| ATP
ADP |
1. |
Pi
H2O |
1) Cholin-Kinase | 2.7.1.32 | Tr | ||
| Mg / Co / Mn | 2) Phosphoethanolamin-/ Phosphocholin-Phosphatase | 3.1.3.75 | Hyd | ||||
|
|||||||
| CTP
PPi |
Cholin-phosphat- Cytidylyltransferase | 2.7.7.15 | Tr | ||||
 CDP-Cholin CDP-Cholin
|
|||||||

Das CDP-Cholin wird benötigt für die Biosynthese von Phosphatidylcholin (Lecithin).
Biosynthese von Betain und Abbau zu Glycin
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| Ox. Akz.
Red. Akz. |
PQQ | Cholin-Dehydrogenase | 1.1.99.1 | Ox | ||||||
|
||||||||||
| H2O, FAD | PQQ | Cholin-Dehydrogenase | 1.1.99.1 | Ox | ||||||
|
||||||||||
| L-Homocystein | Zn | Betain--Homocystein- S-Methyltransferase | 2.1.1.5 | Tr | ||||||
|
||||||||||
| H2O, Ox. Akz.
Formaldehyd, Red. Akz. |
FAD | Dimethylglycin-Dehydrogenase | 1.5.99.2 | Ox | DMGDH-Def. | |||||
|
||||||||||
| H2O, O2
H2O2 |
FAD | Sarcosin-Oxidase | 1.5.3.1 | Ox | ||||||
|
Red. Akz. |
oder
|
FMN | oder
Sarcosin-Dehydrogenase |
1.5.99.1 | Ox | Sarcosinämie | ||||


Umkehrung des letzten Schritts:
| ( ⇓ ) | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
|
|||||||
| S-Adenosyl-L-Homocystein | Glycin-N-Methyltransferase | 2.1.1.20 | Tr | GNMT-Def. | |||
Betain agiert als Methylgruppendonor im Methionin-Stoffwechsel zur Rückgewinnung von Methionin aus Homocystein.
Die Glycin-N-Methyltransferase-Reaktion dient wahrscheinlich nicht der Biosynthese von Sarcosin, dem keine physiologische Bedeutung zukommt, sondern der Regulation des S-Adenosyl-L-Methionin/S-Adenosyl-L-Homocystein-Quotienten in Leber und Pankreas. Durch die Reaktion kann die Konzentration des Methylgruppendonors S-Adenosyl-L-Methionin verringert werden, der beim Methionin-Abbau anfällt, wenn sonst keine Verwendung für die Methylgruppen besteht.
Weblinks
Bearbeiten- KEGG: Glycerophospholipid metabolism - Homo sapiens (human)
- KEGG: Glycine, serine and threonine metabolism - Homo sapiens (human)
Sphingolipid-Stoffwechsel
BearbeitenAllgemeines
Bearbeiten |
 |
Sphingolipide spielen v.a. als Membranlipide im Nervensystem eine wichtige Rolle. Auch die Blutgruppenantigene leiten sich von den Sphingolipiden ab. Sie bestehen aus Sphingosin, einem Aminoalkohol, der über eine Doppelbindung mit einem langen Kohlenstoffschwanz verbunden ist, sowie einer Fettsäure, die an der Sphingosin-Aminogruppe hängt. Nach dem Rest am C1-Atom kann man die Spingolipide in verschiedene Gruppen einteilen:
- Ceramid (R = Wasserstoff)
- Sphingophospholipide:
- Ceramid-1-phosphat (R = Phosphat)
- Sphingomyelin (R = Phosphocholin)
- Glycosphingolipide:
- Cerebroside (R = Monosaccharid, Glucose oder Galactose)
- Sulfatid (R = Sulfogalactose)
- Lacto-Serie (R = Oligosaccharid, enthält Lactosylceramid)
- Neo-Lacto-Serie (R = Oligosaccharid, enthält Lactosylceramid)
- Globoside (R = Oligosaccharid)
- Ganglioside (R = N-Acetylneuraminsäure-haltiges verzweigtes Oligosaccharid)
- Cerebroside (R = Monosaccharid, Glucose oder Galactose)
Sphingophospholipide enthalten ein Phosphat (Phosphoceramid) oder ein Phosphocholin (Sphingomyelin) und sind damit näher mit den Phosphoglyceriden verwandt. Innerhalb der Gruppen unterscheiden sich die Sphingolipide besonders durch die Art der Fettsäure(n).
Der Abbau der Sphingolipide erfolgt (überwiegend?) in den Lysosomen.
Biosynthese von Ceramid
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| L-Serin
CoA-SH, CO2 |
Pyridoxal- phosphat | Serin-C-Palmitoyltransferase | 2.3.1.50 | Tr | Hereditäre sensorische Neuropathie I | |||||
|
||||||||||
| NADPH/H+ | 3-Dehydrosphinganin-Reduktase | 1.1.1.102 | Ox | |||||||
|
||||||||||
| Acyl-CoA | 1. |
Fettsäure
H2O |
1) Sphingosin-N-Acyltransferase | 2.3.1.24 | Tr | |||||
| 2) Ceramidase | 3.5.1.23 | Hyd | Farber Lipo- granulomatose | |||||||
| 2) YDC1 (alkalische Phytoceramidase) | 3.5.1.- | Hyd | ||||||||
|
||||||||||
| O2, NADPH/H+ ?
2 H2O, NADP+ ? |
DEGS2 (degenerative spermatocyte homolog 2, lipid desaturase (Drosophila)) | 1.14.-.- | Ox | |||||||
|
||||||||||




Ceramid ist die Ausgangssubstanz für das Sphingomyelin und für die Glycosphingolipide.
Erzeugung von Dihydrosphingosin-1-phosphat aus Dihydrosphingosin (Sphinganin)
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| ATP
ADP |
1. |
Pi
H2O |
1) Sphinganin-Kinase | 2.7.1.91 | Tr | |||||
| 2) Phosphatidat-Phosphatase | 3.1.3.4 | Hyd | ||||||||
| 2) Sphingosin-1-phosphat-Phosphatase | 3.1.3.- | Hyd | ||||||||
|
||||||||||

Bildung von Sphingosin und Sphingosin-1-phosphat aus Ceramid
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| H2O | Ceramidase | 3.5.1.23 | Hyd | Farber Lipo- granulomatose | ||||||
|
||||||||||
| ATP
ADP |
1. |
Pi
H2O |
1) Sphinganin-Kinase | 2.7.1.91 | Tr | |||||
| 2) Phosphatidat-Phosphatase | 3.1.3.4 | Hyd | ||||||||
| 2) Sphingosin-1-phosphat-Phosphatase | 3.1.3.- | Hyd | ||||||||
|
||||||||||


Sphingophospholipide: Bildung von Ceramid-1-phosphat aus Ceramid
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| ATP
ADP |
1. |
Pi
H2O |
1) Ceramid-Kinase | 2.7.1.138 | Tr | |||||
| 2) Phosphatidat-Phosphatase | 3.1.3.4 | Hyd | ||||||||
| 2) Sphingosin-1-phosphat-Phosphatase | 3.1.3.- | Hyd | ||||||||
|
||||||||||

Sphingophospholipide: Biosynthese von Sphingomyelin aus Ceramid
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| CDP-Cholin | 1. |
Cholinphosphat
H2O |
1) Ceramid- Cholinphosphotransferase | 2.7.8.3 | Tr | |||||
| 2) Sphingomyelin- Phosphodiesterase | 3.1.4.12 | Hyd | Niemann-Pick-Krankheit A, B | |||||||
|
||||||||||

Sphingomyelin findet man insbesondere in den Myelinscheiden des Nervensystems.
Cerebroside und Derivate: Galactosylceramide
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| UDP-Galactose
UDP |
1. |
Galactose
H2O |
1) N-Acylsphingosin- Galactosyltransferase | 2.4.1.47 | Tr | |||||
| 2) Galactosylceramidase | 3.2.1.46 | Hyd | Morbus Krabbe | |||||||
|
||||||||||
| Galactose
H2O |
Mg, NAD | α-Galactosidase | 3.2.1.22 | Hyd | Morbus Fabry | |||||
|
||||||||||
| 3'-Phosphoadenylylsulfat (PAPS)
Adenosin-3',5'-bisphosphat (PAP) |
Galactosylceramid-Sulfotransferase | 2.8.2.11 | Tr | |||||||
|
||||||||||



Auch D-Galactosylceramid kann sulfatiert werden:
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| 3'-Phosphoadenylylsulfat (PAPS)
Adenosin-3',5'-bisphosphat (PAP) |
1. |
Sulfat
H2O |
1) Galactosylceramid- Sulfotransferase | 2.8.2.11 | Tr | |||||
| 2) Arylsulfatase | 3.1.6.1 | Hyd | ||||||||
| 2) Cerebrosid-Sulfatase | 3.1.6.8 | Hyd | Metachromatische Leukodystrophie | |||||||
|
||||||||||

Abbau von GM4 (N-Acetylneuraminyl-galactosylceramid) zu Galactosylceramid:
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| H2O | Exo-α-Sialidase (α-Neuraminidase) | 3.2.1.18 | Hyd | Sialidose II (Mucolipidose I) | ||||||
|
||||||||||

Cerebroside und Derivate: Glucosylceramide
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| UDP-Glucose
UDP |
1. |
Glucose
H2O |
1) Ceramid-Glucosyltransferase | 2.4.1.80 | Tr | |||||
| 2) Glucosylceramidase (saure β-Glucosidase) | 3.2.1.45 | Hyd | Morbus Gaucher Typ I, II, III, IIIc, neonatal letal | |||||||
|
||||||||||
| UDP-Galactose
UDP |
1. |
Galactose
H2O |
1) β-1,4-Galactosyltransferase | 2.4.1.- | Tr | |||||
| 2) β-Galactosidase | 3.2.1.23 | Hyd | GM1-Gangliosidose, Mucopolysaccharidose IVb (Morquio B) | |||||||
|
||||||||||


Lactosylceramid ist der Ausgangspunkt für die Biosynthese der verschiedenen Glycosphingolipide, z.B. der Blutgruppen-Antigene.
Weblinks
Bearbeiten
Ketonkörperbiosynthese
BearbeitenAllgemeines
BearbeitenDie Ketonkörper Acetoacetat und β-Hydroxybutyrat werden besonders in Fastenzeiten in der Leber gebildet und dienen als alternative Energieträger.
Biosynthese von Acetoacetat und β-Hydroxybutyrat aus Acetyl-CoA
Bearbeiten| Tr. | ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||
| Acetyl-CoA | Acetyl-CoA | Acetyl-CoA-C-Acetyltransferase Mitochondrium | 2.3.1.9 | Tr | α-Methylaceto- acetacidurie | ||||||
|
|||||||||||
| + SRE-1 | H2O, Acetyl-CoA | HMG-CoA-Synthase Mitochondrium | 2.3.3.10 | Tr | HMGCS2-Def. | ||||||
|
|||||||||||
| HMG-CoA-Lyase | 4.1.3.4 | Ly | HMGCL-Def. | ||||||||
|
|||||||||||
| NADH/H+ | NADH/H+ | β-Hydroxybutyrat-Dehydrogenase Mitochondrium | 1.1.1.30 | Ox | |||||||
|
|||||||||||
Die Biosynthese von HMG-CoA steht auch am Anfang der Cholesterin-Biosynthese.
Acetoacetat entsteht auch beim Abbau der Aminosäuren Phenylalanin/Tyrosin, HMG-CoA fällt beim Leucin-Abbau an.
Spontaner Zerfall von Acetoacetat zu Aceton
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| H+
CO2 |
spontan | |||||||||
|
||||||||||
Reaktivierung von Acetoacetat zu Acetoacetyl-CoA und Rückgewinnung von 2 Acetyl-CoA
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| Succinyl-CoA | Succinyl-CoA | Acetoacetat-Succinyl-CoA- Transferase Mitochondrium | 2.8.3.5 | Tr | SCOT-Def. | |||||
|
||||||||||
| CoA-SH
|
|
CoA-SH
|
Acetyl-CoA-C-Acyltransferase (β-Ketothiolase) | 2.3.1.16 | Tr | TFP-Def. | ||||
| Acetyl-CoA-C-Acetyltransferase (Acetoacetyl-CoA-Thiolase) | 2.3.1.9 | α-Methylacetoacetacidurie | ||||||||
|
||||||||||

Biologische Bedeutung
BearbeitenBei längeren Fastenzeiten greift der Körper auf seine Fettreserven zurück (Hemmung der Lipogenese, Stimulation der Lipolyse). Die ans Blut abgegebenen freien Fettsäuren können jedoch nicht von allen Organen verbrannt werden. Auch die Gluconeogenese ist aus Fettsäuren nicht möglich, was insbesondere für Glucose-abhängige Organe problematisch ist. In der Leber werden aus Fettsäuren bzw. dem vermehrt anfallenden Acetyl-CoA nun Ketonkörper gebildet, die als alternative Energieträger dienen. Das Gehirn, das normalerweise überwiegend von Glucose lebt, kann sich so z.B. bei längerer Nahrungskarenz umstellen und dann einen Großteil seines Energiebedarfs über Ketonkörper decken.
Die Ketonkörper Acetacetat und β-Hydroxybuttersäure werden (nur) von der Leber produziert, aber nicht von ihr verwertet. In der Peripherie können die alternativen Energieträger wieder mit Coenzym A aktiviert werden (reversible Übertragung des CoA von Succinyl-CoA auf Acetacetat) und im letzten Schritt der β-Oxidation der Fettsäuren (β-Ketothiolase) zu Acetyl-CoA zerlegt werden. Aceton wird nicht nennenswert verwertet.
Pathobiochemie
BearbeitenBei der Ketoazidose des Typ-I-Diabetikers führt der absolute Insulinmangel zum intrazellulären Glucose- und damit Energiemangel. Als Reaktion darauf wird die Lipolyse aktiviert und die erhöhte Serumkonzentration an freien Fettsäuren fließt in die hepatische Ketonkörpersynthese. Neben der Azidose lassen sich die Ketonkörper im Urin nachweisen, das übermäßig gebildete Aceton führt zum typischen süßlich-obstartigen Foetor ex ore.
Weblinks
Bearbeiten- KEGG: Synthesis and degradation of ketone bodies - Reference pathway
- KEGG: Butanoate metabolism - Reference pathway
Cholesterinbiosynthese
BearbeitenAllgemeines
BearbeitenCholesterin findet sich in Zellmembranen. Im Intermediärstoffwechsel ist es der Rohstoff für die Biosynthese von Steroidhormonen und Gallensäuren. Vom Cholesterin-Biosyntheseweg zweigen auch die Synthesewege von Ubichinon (Coenzym Q), Dolichol-Phosphat und Cholecalciferol (Vitamin D-Hormon) ab.
Biosynthese von „aktivem Isopren“ aus Acetyl-CoA
Bearbeiten| Tr. | ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||
| Acetyl-CoA | Acetyl-CoA | Acetyl-CoA C-Acetyltransferase Mitochondrium | 2.3.1.9 | Tr | α-Methylaceto- acetacidurie | ||||||
|
|||||||||||
| + SRE-1 | H2O, Acetyl-CoA | HMG-CoA-Synthase Mitochondrium | 2.3.3.10 | Tr | HMGCS2-Def. | ||||||
|
|||||||||||
| + SRE-1 | 2 NADPH/H+ | HMG-CoA-Reduktase Zytosol | 1.1.1.34 | Ox | |||||||
|
|||||||||||
| ATP
ADP |
Mevalonat-Kinase
Peroxisom |
2.7.1.36 | Tr | Hyper-IgD-S., Mevalon- acidurie | |||||||
|
|||||||||||
| ATP
ADP |
Phosphomevalonatkinase Peroxisom | 2.7.4.2 | Tr | ||||||||
|
|||||||||||
| ATP
ADP |
Pyrophosphomevalonat- Decarboxylase Peroxisom | 4.1.1.33 | Ly | ||||||||
|
|||||||||||
|
|
|||||||||||
|
|||||||||||
| FMN / FAD; Mg / Mn / Ca | 3-Isopentenyl- pyrophosphat-Isomerase | 5.3.3.2 | Iso | ||||||||
|
|||||||||||
Der erste Schritt, die Bildung von Acetoacetyl-CoA aus 2 Acetyl-CoA entspricht der Umkehrung des letzten Schritts der β-Oxidation.
Terpenoid-Biosynthese
BearbeitenTerpenoid-Biosynthese durch Zusammenbau von 3 Isopreneinheiten zum Farnesylpyrophosphat. Kopf-Kopf-Kondensation von 2 Farnesylpyrophosphaten zum Squalen:
| Tr. | ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||
| + SRE-1 | Isopentenyl-PP
PPi |
Dimethylallyl-transtransferase | 2.5.1.1 | Tr | |||||||
| + SRE-1 | Geranyltranstransferase | 2.5.1.10 | Tr | ||||||||
|
|||||||||||
| + SRE-1 | Isopentenyl-PP
PPi |
Dimethylallyl- transtransferase | 2.5.1.1 | Tr | |||||||
| + SRE-1 | Geranyltranstransferase | 2.5.1.10 | Tr | ||||||||
|
|||||||||||
|
|
Mg / Mn | Squalen-Synthase | 2.5.1.21 | Tr | |||||||
|
|||||||||||
| 2 NADPH/H+
PPi, 2 NADP+ |
|||||||||||
|
|||||||||||


Ringschluß und Modifikation der Ringe und Seitenketten
BearbeitenRingschluß von Squalen zum Viererring-System und Modifikation der Ringe und Seitenkette zum Cholesterin:
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| AH2, O2
A, H2O |
FAD | Squalen-Monooxygenase | 1.14.99.7 | Ox | ||||||
|
||||||||||
| Lanosterol-Synthase | 5.4.99.7 | Iso | ||||||||
|
||||||||||
| 3 O2, 3 NADPH/H+ | Häm-Thiolat | Sterol-14-Demethylase | 1.14.13.70 | Ox | ||||||
|
||||||||||
| NADPH/H+ | δ-14-Sterol-Reduktase | 1.3.1.70 | Ox | HEM-Skelett- dyplasie | ||||||
|
||||||||||
| 3 O2, 3 NADPH/H+
4 H2O, 3 NADP+ |
Methylsterol- Monooxygenase | 1.14.13.72 | Ox | |||||||
|
||||||||||
| NADP+ | Sterol-4-α-carboxylat- 3-Dehydrogenase (decarboxylierend) | 1.1.1.170 | Ox | CHILD-Syndrom | ||||||
|
||||||||||
| NADPH/H+ | 3-Keto-Steroid-Reduktase | 1.1.1.270 | Ox | |||||||
|
||||||||||
| ? [H]
? [CH3] |
multi-step reaction | |||||||||
|
||||||||||
| Cholestenol-δ-Isomerase * | 5.3.3.5 | Iso | Conradi-Hunermann-Happle-S. | |||||||
|
||||||||||
| NADPH/H+ | NADPH/H+ | δ-24-Sterol-Reduktase * | 1.3.1.- | Ox | Desmosterolosis | |||||
|
||||||||||
| O2, NADH/H+
2 H2O, NADP+ |
Lathosterol-5-Desaturase | 1.14.21.6 | Ox | Lathosterolosis | ||||||
|
||||||||||
| NADH/H+ | 7-Dehydrocholesterol- Reduktase | 1.3.1.21 | Ox | Smith-Lemli-Opitz-S. (SLOS) | ||||||
|
||||||||||
-Squalen-2,3-epoxid.svg)











 |
* Umgekehrte Reihenfolge möglich. Zwischenprodukt ist dann nicht 5α-Cholesta-7,24-dien-3β-ol, sondern Zymostenol.
Squalen wird zuerst in das reaktionsfreudige Squalen-Epoxid umgewandelt. Danach erfolgt der Zusammenschluß der 4 Ringe, die das Steroid-Gerüst bilden. Vom entstandenen Lanosterol bis zum fertigen Cholesterinmolekül folgen nun diverse Modifikationen:
- Sukzessive Beseitigung von 3 Methylgruppen (zwei in Position 4 und eine in Position 14)
- Verlagerung der Doppelbindung von Position 8 nach Position 5 über mehrere Zwischenschritte
- Auflösung der Doppelbindung in der Seitenkette
Das Steran-(Gonan-)Ringgerüst ist aus vier homozyklischen Kohlenstoffringen zusammengesetzt und besteht aus 17 C-Atomen. Mit der Seitenkette sind es 26 C-Atome.
Stoff- und Energiebilanz
BearbeitenZur Synthese eines Cholesterinmoleküls werden sechs HMG-CoA bzw. 18 Acetyl-CoA benötigt. Außerdem kostet die Synthese eine Menge Energie, nämlich 25 Reduktionsäquivalente (überwiegend NADPH/H+) und 18 ATP.
Für die Bildung eines Cholesterin-Moleküls müssen daher netto ca. 10 bis 11 Glucose-Moleküle abgebaut werden: 9 für die Bildung von 18 Acetyl-CoA (dabei werden bereits die benötigten 18 ATP gebildet) und mind. 2 Glucose-Moleküle für die NADPH/H+-Produktion im HMP-Weg (alternativ kann NADPH/H+ im Citrat-Malat-Pyruvat-Zyklus aus NADH/H+ erzeugt werden). Etwas positiver wird die Bilanz, wenn man noch die 18 NADH/H+ (entspr. 45 ATP oder dem vollständigen Abbau von 1,4 Glucose-Molekülen) gegenrechnet, die bei der dehydrierenden Decarboxylierung der 9 Glucose-Moleküle gewonnen werden.
Biologische Bedeutung
BearbeitenCholesterin wird in Zellmembranen eingebaut und ist der Rohstoff für die Biosynthese der Steroidhormone wie Aldosteron, Cortisol und Sexualsteroide. Das Cholesterinvorläufermolekül 7-Dehydrocholesterol dient auch der Biosynthese von Vitamin D-Hormon. Im Blut wird Cholesterin v.a. als Cholesterinester in low density lipoproteins (LDL) transportiert. LDL transportiert Cholesterin vorwiegend von der Leber in die Peripherie, während high density lipoproteins (HDL) Cholesterin in die Gegenrichtung transportieren.
Regulation
BearbeitenDie Cholesterinsynthese erfolgt bedarfsorientiert und in Abhängigkeit vom Angebot in der Nahrung, reguliert durch das Sterolregulationselement 1 (SRE-1). Daraus ergibt sich, dass sich der Cholesterinspiegel über diätetische Maßnahmen nur gering beeinflussen lässt. Die Elimination erfolgt durch die Umwandlung in Gallensäuren.
Verbindungen zu anderen Stoffwechselwegen
Bearbeiten- Die Bildung von β-Hydroxy-β-methylglutaryl-CoA (HMG-CoA) bildet auch den Start der Ketonkörperbiosynthese.
- Vom Farnesylpyrophosphat zweigt die Biosynthese von Dolicholphosphat und Coenzym Q ab.
- Aus 7-Dehydrocholesterol wird Vitamin-D-Hormon gebildet.
- Cholesterin selbst ist der Ausgangspunkt für die Biosynthese der Steroidhormone und Gallensäuren.
Pathobiochemie
BearbeitenEin hohes LDL in Kombination mit einem niedrigen HDL fördert die Atherosklerose der Gefäße. Ein genetischer Defekt des LDL-Rezeptors führt zum Krankheitsbild der familiären Hypercholesterinämie (OMIM), bei der es frühzeitig zu einer schweren Atherosklerose kommt.
Pathologie
BearbeitenHistologisch finden sich in Atheroskleroseplaques neben lipidbeladenen Makrophagen (Schaumzellen) häufig typische spaltförmige Cholesterin-Lücken (cholesterol clefts, cholesterol imprints), die dadurch entstehen, dass die Cholesterinkristalle bei der Vorbehandlung ausgewaschen werden.
Pharmakologie
BearbeitenDie HMG-CoA-Reduktase ist das pharmakologische Target der Statine (CSE-Hemmer), die bei Hypercholesterinämie und zur Progressionsverzögerung der Atherosklerose eingesetzt werden. Von der Cholesterin-Synthese zweigt auch auf Höhe des Zymosterols die Ergosterol-Synthese der Pilze ab. Daher können mit Cholesterinsynthese-Hemmstoffen, die vor dieser Abzweigung ansetzen, auch Pilzinfektionen behandelt werden. Beispiele für derartige Antimykotika sind Amorolfin, Naftifin und Terbinafin, die die Squalen-Monooxygenase (EC 1.14.99.7) hemmen und Azol-Antimykotika wie z.B. Clotrimazol, die die Sterol-14-Demethylase (EC 1.14.13.70) inhibieren.
Weblinks
Bearbeiten
Vitamin D-Stoffwechsel
BearbeitenAllgemeines
BearbeitenVitamin D-Hormon fördert die Kalziumresorption und darüber die Knochenmineralisierung. Die Vorstufe Vitamin D3 wird bei Sonneneinstrahlung in der Haut gebildet und/oder über die Nahrung aufgenommen.
Biosynthese
Bearbeiten| Tr. | ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||
|
hν ~ |
|||||||||||
|
|||||||||||
| O2, NADPH/H+
H2O, NADP+ |
Häm-Thiolat | Vitamin-D–25-Hydroxylase (CYP2R1) Leber | 1.14.15.15 | Ox | VDDR 1B | ||||||
|
|||||||||||
| + PTH + PRL |
O2, NADPH/H+
H2O, NADP+ |
Häm-Thiolat | Calcidiol-1-Monooxygenase (Cyp27B1) Niere |
1.14.15.18 | Ox | VDDR 1A | |||||
|
|||||||||||



Vitamin D-Hormon ist eigentlich kein echtes Vitamin, da es im Körper selbst aus der Cholesterin-Vorstufe 7-Dehydrocholesterol gebildet wird. Das Ringsystem von 7-Dehydrocholesterol, das vor allem in der Leber synthetisiert wird, wird in der Haut durch UV-Licht gesprengt, so dass Cholecalciferol (Vitamin D3) entsteht. Vitamin D3 wird auch aus der Nahrung aufgenommen. Dieses wird nun in der Leber zu 25-Hydroxycholecalciferol und bei Bedarf unter Einfluss von Parathormon (PTH) in der Niere zum aktiven 1,25-Dihydroxycholecalciferol (D-Hormon, Calcitriol) hydroxyliert.
Der größte Teil des Vitamins wird bei ausreichender Lichtexposition im Körper selbst gebildet. In Nahrungsmitteln kommt es in nennenswerten Mengen nur in tierischen Produkten vor (Fisch, Eigelb, Milchprodukte, Rinderleber). Pflanzliche Lebensmittel enthalten meist nur das Provitamin Ergosterol, eine Vorstufe des Vitamin D2.
Ausscheidung
BearbeitenWie bei allen anderen Steroiden (lipophile Moleküle!) erfolgt die Ausscheidung vorwiegend über die Leber.
Wirkung
BearbeitenWie viele andere lipophile Signalstoffe (z.B. Steroidhormone) wirkt Calcitriol über die Bindung an intrazelluläre Rezeptoren, die die Genexpression beeinflussen. Calcitriol fördert die Kalzium- und Phosphatresorption im Darm und die Kalzium- und Phosphatrückresorption in der Niere und ist somit an der Konstanthaltung des Serum-Kalziumspiegels und an der Knochenmineralisierung beteiligt. Es spielt auch eine Rolle bei der Differenzierung und Reifung der Immunzellen. Weiterhin gibt es Hinweise für eine tumorprotektive Wirkung.
Mangelerkrankungen
BearbeitenEin beginnender Mangel manifestiert sich in brüchigen Fingernägeln, unter den Nägeln bilden sich mehrere weiße Flecken. Rachitis bei Kindern, Osteomalazie bei Erwachsenen. Ein Calcitriol-Mangel ist zu befürchten bei völlig fehlender Sonnenexposition und bei Niereninsuffizienz.
sekundärer Hyperparathyreoidismus
Weblinks
Bearbeiten- KEGG: Biosynthesis of steroids - Homo sapiens (human)
- RCSB PDB: Vitamin D Receptor
- Cholecalciferol Mangel
Gallensäuren-Stoffwechsel
BearbeitenAllgemeines
BearbeitenZu den Gallensäuren gehören die Cholsäure, die Taurocholsäure, die Glycocholsäure, die Chenodesoxycholsäure und die Desoxycholsäure. Sie werden aus Cholesterin gebildet. Im Darm helfen sie bei der Fettverdauung und -resorption.
Biosynthese von 7α-Hydroxycholest-4-en-3-on, dem Startpunkt der Gallensäuren-Biosynthese
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| H2O | 1. |
CoA-SH | 1) Sterol-Esterase | 3.1.1.13 | Hyd | Wolman-Krankheit | ||||
| 2) Sterol-O-Acyltransferase | 2.3.1.26 | Tr | ||||||||
|
||||||||||
| O2, NADPH/H+
H2O, NADP+ |
Häm- Thiolat | Cholesterol-7α- Monooxygenase | 1.14.13.17 | Ox | ||||||
|
||||||||||
| NADP+ | Cholest-5-en-3β,7α-diol - 3β-Dehydrogenase | 1.1.1.181 | Ox | |||||||
|
||||||||||



Ab dem 7α-Hydroxycholest-4-en-3-on verzweigt sich die Gallensäurenbiosynthese in zwei längere Wege, die sich grob betrachtet nur in der ersten Reaktion - einer zusätzlichen Hydroxylierung in der 12α-Position - unterscheiden. Der erste Weg führt zum Glycocholat, Taurocholat und Cholat. Der zweite zum Chenodeoxyglycocholat und Chenodeoxycholat.
Biosynthese von Choloyl-CoA
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| O2, NADPH/H+
H2O, NADP+ |
Oxidoreduktase | 1.14.13.- | Ox | |||||||
|
||||||||||
| NADPH/H+ | 3-oxo-5β-steroid-4-Dehydrogenase | 1.3.99.6 | Ox | |||||||
|
||||||||||
| NADH/H+ | Cholest-5-en-3β,7α-diol-3β-Dehydrogenase | 1.1.1.50 | Ox | |||||||
|
||||||||||
| O2, NADPH/H+
H2O, NADP+ |
Häm- Thiolat | Cholestantriol-26-Monooxygenase | 1.14.13.15 | Ox | CTX | |||||
|
||||||||||
| NAD+ | Cholestanetetraol- 6-Dehydrogenase | 1.1.1.161 | Ox | |||||||
|
||||||||||
| H2O, NAD+ | 3α,7α,12α-Trihydroxycholestan-26-al-26-Oxidoreductase | 1.2.1.40 | Ox | |||||||
|
||||||||||
| CoA-SH, ATP
AMP, PPi |
Mg | Cholat--CoA-Ligase | 6.2.1.7 | Lig | ||||||
|
||||||||||
| FAD | Oxidoreduktase (Acyl-Coenzym A-Dehydrogenase-Familie) | 1.3.99.- | Ox | |||||||
|
||||||||||
| H2O
|
Hydro-lyase | 4.2.1.- | Ly | |||||||
|
||||||||||
| NAD+ | Oxidoreduktase | 1.1.1.- | Ox | |||||||
|
||||||||||
| CoA-SH | Propanoyl-CoA C-Acyltransferase | 2.3.1.176 | Tr | Zellweger- Syn. | ||||||
|
||||||||||
| AMP, PPi
CoA-SH, ATP |
AMP, PPi
ATP, CoA-SH |
Mg | Cholat--CoA-Ligase | 6.2.1.7 | Lig | |||||
|
||||||||||
| ?
? |
||||||||||
|
||||||||||













Aus Choloyl-CoA werden Taurocholat und Glycocholat
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
|
Gallensäure-CoA:Aminosäure-N-Acyltransferase | 2.3.1.65 | Tr | Familiäre Hypercholanämie | ||||||
|
||||||||||

| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
|
Gallensäure-CoA:Aminosäure-N-Acyltransferase | 2.3.1.65 | Tr | Familiäre Hypercholanämie | ||||||
|
||||||||||

Zusammenfassung: Zuerst wird das Vierer-Ring-System Cholesterin mit insgesamt drei Hydroxyl-Gruppen versehen und die Doppelbindung wird entfernt. Danach wird das Ende der Seitenkette oxidiert, klassisch vom Alkan- zum Alkohol-, über einen Aldehyd- zum Carbonsäure-Rest. Die gebildete Carboxyl-Gruppe kann nun unter ATP-Verbrauch mit Coenzym A aktiviert werden. Nun wird die Seitenkette um drei C-Atome (inklusive der Methyl-Gruppe) gekürzt nach dem Muster der β-Oxidation. Die Seitenkette des entstandenen Choloyl-CoA wird nun mit Taurin (Taurocholat) oder Glycin (Glycocholat) über eine Säureamid-Bindung verbunden oder das Coenzym A wird einfach abgespalten (die Auflösung der energiereichen Thioester-Bindung wird zur Bildung eines ATPs genutzt) und es entsteht Cholat.
Biosynthese von Chenodeoxyglycocholat
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| NADPH/H+ | 3-oxo-5β-steroid- 4-Dehydrogenase | 1.3.99.6 | Ox | |||||||
|
||||||||||
| NADH/H+ | Cholest-5-en-3β,7α-diol-3β-Dehydrogenase | 1.1.1.50 | Ox | |||||||
|
||||||||||
| O2, NADPH/H+
H2O, NADP+ |
Häm- Thiolat | Cholestantriol-26-Monooxygenase | 1.14.13.15 | Ox | CTX | |||||
|
||||||||||
| NAD+ | Zn / Fe | Alkohol-Dehydrogenase (NAD+) | 1.1.1.1 | Ox | ||||||
|
||||||||||
| H2O, NAD+ | Aldehyddehydrogenase (NAD+) | 1.2.1.3 | Ox | Alk.int., SLS | ||||||
|
||||||||||
| CoA-SH, ATP
AMP, PPi |
Mg | Cholat--CoA-Ligase | 6.2.1.7 | Lig | ||||||
|
||||||||||
| FAD | Oxidoreduktase (Acyl-Coenzym A-Dehydrogenase-Familie) | 1.3.99.- | Ox | |||||||
|
||||||||||
| H2O
|
Hydro-lyase | 4.2.1.- | Ly | |||||||
|
||||||||||
| NAD+ | Oxidoreduktase | 1.1.1.- | Ox | |||||||
|
||||||||||
| CoA-SH | Acetyl-CoA-C-Acyltransferase (β-Ketothiolase) | 2.3.1.16 | Tr | TFP-Def. | ||||||
|
||||||||||
| AMP, PPi
CoA-SH, ATP |
Mg | Cholat--CoA-Ligase | 6.2.1.7 | Lig | ||||||
|
||||||||||
| ?
? |
||||||||||
|
||||||||||












Eine Abkürzung: Vom 3α,7α-Dihydroxy-5β-cholestanoyl-CoA direkt zum Chenodeoxycholoyl-CoA
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| CoA-SH | Acetyl-CoA-C-Acyltransferase (β-Ketothiolase) | 2.3.1.16 | Tr | TFP-Def. | ||||||
|
||||||||||
Aus Chenodeoxycholoyl-CoA wird Chenodeoxyglycocholat
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
|
Gallensäure-CoA:Aminosäure-N-Acyltransferase | 2.3.1.65 | Tr | Familiäre Hypercholanämie | ||||||
|
||||||||||

Die Biosynthese von Chenodeoxyglycocholat verhält sich analog zur Biosynthese von Glycocholat (s.o.), es fehlt nur die anfängliche Hydroxylierung in der 12α-Position.
Aufgaben der Gallensäuren
BearbeitenGallensäuren werden in der Leber gebildet und aus dem Pfortaderkreislauf resorbiert. In die Galle gelangen sie durch aktive Sekretion.
Als Emulgatoren unterstützen die amphiphilen Gallensäuren im Darm die Fettverdauung durch Lösung der Fette im wässrigen Milieu. Dadurch erhöht sich die Angriffsfläche für die Lipasen. Im Ileum werden 98 % der Gallensalze zurückresorbiert (enterohepatischer Kreislauf). Von einer funktionierenden Fettresorption hängt auch die Resorption der fettlöslichen Vitamine A, E, D, K (Merke: EDEKA) ab.
In der Galle wirken die Gallensäuren ebenfalls als Lösungsvermittler und zwar für das lipophile Cholesterin. Zusammen mit Phospholipiden wirken sie dort der Entstehung von (Cholesterin-)Gallensteinen entgegen.
Weblinks
Bearbeiten
Steroidhormon-Stoffwechsel
BearbeitenAllgemeines
BearbeitenDie lipophilen Steroidhormone werden überwiegend in der Nebennierenrinde und den Gonaden aus Cholesterol (Cholesterin) gebildet. Die Synthese erfolgt zeitgleich zur Stimulation, da die Speicherkapazitäten gering sind. Metabolisiert werden sie größtenteils in der Leber über Enzyme der Cytochrom-P450-Familie, z.T. auch in anderen Organen (Umwandlung von Androgenen in Östrogene im Fettgewebe durch eine Aromatase). Wie an den EC-Nummern zu sehen ist handelt es sich dabei größtenteils um Oxidoreduktase-Reaktionen (Hydroxylase-Reaktionen) am Steroidgerüst. Ausgeschieden werden sie vorwiegend als Glucuronide über die Leber.
Hinweis: Zur besseren Nachvollziehbarkeit der Auswirkungen der verschiedenen Enzymdefekte wurden die entsprechenden Enzyme in den folgenden Tabellen farblich unterlegt. Die Darstellung ist auf das Wesentliche reduziert, weitere Stoffwechselschritte und Metabolite findet man bei KEGG (siehe Weblinks).
Biosynthese von Pregnenolon, Progesteron und 11β-Hydroxyprogesteron aus Cholesterol
BearbeitenCholesterol ⇓ 1.14.15.6 20α-OH-Cholesterol/ 22β-OH-Cholesterol ⇓ 1.14.15.6 20α,22β-Dihydroxy- cholesterol ⇓ 1.14.15.6 Pregnenolon ⇓ ⇑ 1.1.1.145 / 5.3.3.1 Progesteron ⇓ 1.14.15.4 11β-OH-Progesteron
Reaktionen
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||
| O2, NADPH/H+
H2O, NADP+ |
Häm- Thiolat | Cholesterol-Monooxygenase (side-chain-cleaving) Cholesterol-20-22-Desmolase | 1.14.15.6 | Ox | (AGS I durch STAR-Def.) | ||||||
|
|||||||||||
| O2, NADPH/H+
H2O, NADP+ |
Häm- Thiolat | Cholesterol-Monooxygenase (side-chain-cleaving) Cholesterol-20-22-Desmolase | 1.14.15.6 | Ox | (AGS I durch STAR-Def.) | ||||||
|
|||||||||||
| Ox. Ferredoxin
Red. Ferredoxin, 4-Methylpentanal |
Häm- Thiolat | Cholesterol-Monooxygenase (side-chain-cleaving) Cholesterol-20-22-Desmolase | 1.14.15.6 | Ox | (AGS I durch STAR-Def.) | ||||||
|
|||||||||||
| NAD+ | NAD+ | 3β-Hydroxy-δ5-Steroid- Dehydrogenase | 1.1.1.145 | Ox | AGS II | ||||||
| Steroid-δ-Isomerase | 5.3.3.1 | Iso | |||||||||
|
|||||||||||
| O2, Red. Ferredoxin
H2O, Ox. Ferredoxin |
Häm- Thiolat | Steroid-11β-Monooxygenase (Steroid-11β-Hydroxylase) | 1.14.15.4 | Ox | AGS IV | ||||||
|
|||||||||||





Biosynthese der Mineralokortikoide
BearbeitenPregnenolon ⇒ 21-OH-Pregnenolon
1.14.99.10
⇓ ⇑ 1.1.1.145 / ⇓ 1.1.1.145 /
5.3.3.1 5.3.3.1
Progesteron ⇒ 11-Deoxycorticosteron
1.14.99.10
⇓ 1.14.15.4 ⇓ 1.14.15.4
11β-OH-Progesteron ⇒ Corticosteron ⇒ 18-OH-Corticosteron ⇒ Aldosteron
1.14.99.10 1.14.15.5
Reaktionen
2. Spalte (waagerecht)
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| O2, Red. Akz.
H2O, Ox. Akz. |
Häm-Thiolat | Steroid-21-Monooxygenase (Steroid-21-Hydroxylase) | 1.14.99.10 | Ox | AGS III | |||||
|
||||||||||

| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| O2, Red. Akz.
H2O, Ox. Akz. |
Häm-Thiolat | Steroid-21-Monooxygenase (Steroid-21-Hydroxylase) | 1.14.99.10 | Ox | AGS III | |||||
|
||||||||||

| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| O2, Red. Akz.
H2O, Ox. Akz. |
Häm-Thiolat | Steroid-21-Monooxygenase (Steroid-21-Hydroxylase) | 1.14.99.10 | Ox | AGS III | |||||
|
||||||||||

3. Spalte (senkrecht)
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| NAD(P)+ | 3β-Hydroxy-δ5-Steroid- Dehydrogenase | 1.1.1.145 | Ox | AGS II | ||||||
| Steroid-δ-Isomerase | 5.3.3.1 | Iso | ||||||||
|
||||||||||
| O2, Red. Ferredoxin
H2O, Ox. Ferredoxin |
Häm- Thiolat | Steroid-11β-Monooxygenase (Steroid-11β-Hydroxylase) | 1.14.15.4 | Ox | AGS IV | |||||
|
||||||||||
4. Spalte (waagerecht)
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| O2, Red. Ferredoxin
H2O, Ox. Ferredoxin |
Häm-Thiolat | Corticosteron-18-Monooxygenase (Corticosteron-18-Hydroxylase) | 1.14.15.5 | Ox | Aldosteron-Def. I, Typ II | |||||
|
||||||||||
| Ox. Akzeptor (?)
Red. Akzeptor (?) |
? | |||||||||
|
||||||||||

Biosynthese der Glukokortikoide
BearbeitenPregnenolon ⇒ 17α-OH-Pregnenolon ⇒ 17α,21-Dihydroxypregnenolon
1.14.99.9 1.14.99.10
⇓ ⇑ 1.1.1.145 / ⇓ ⇑ 1.1.1.145 / ⇓ 1.1.1.145 /
5.3.3.1 5.3.3.1 5.3.3.1
Progesteron ⇒ 17α-OH-Progesteron ⇒ 11-Deoxycortisol
1.14.99.9 1.14.99.10
⇓ 1.14.15.4 ⇓ 1.14.15.4 ⇓ 1.14.15.4
11β-OH-Progesteron ⇒ 21-Deoxycortisol ⇒ Cortisol ⇒ Cortison
1.14.99.9 1.14.99.10 1.1.1.146
Reaktionen
2. Spalte (waagerecht)
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| O2, Red. Akz.
H2O, Ox. Akz. |
Häm-Thiolat | Steroid-17α-Monooxygenase (Steroid-17α-Hydroxylase) | 1.14.99.9 | Ox | AGS V | |||||
|
||||||||||

| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| O2, Red. Akz.
H2O, Ox. Akz. |
Häm-Thiolat | Steroid-17α-Monooxygenase (Steroid-17α-Hydroxylase) | 1.14.99.9 | Ox | AGS V | |||||
|
||||||||||

| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| O2, Red. Akz.
H2O, Ox. Akz. |
Häm-Thiolat | Steroid-17α-Monooxygenase (Steroid-17α-Hydroxylase) | 1.14.99.9 | Ox | AGS V | |||||
|
||||||||||

3. Spalte (senkrecht)
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| NAD(P)+ | NAD(P)+ | 3β-Hydroxy-δ5-Steroid- Dehydrogenase | 1.1.1.145 | Ox | AGS II | |||||
| Steroid-δ-Isomerase | 5.3.3.1 | Iso | ||||||||
|
||||||||||
| O2, NADPH/H+
H2O, NADP+ |
Häm- Thiolat | Steroid-11β-Monooxygenase (Steroid-11β-Hydroxylase) | 1.14.15.4 | Ox | AGS IV | |||||
|
||||||||||
4. Spalte (waagerecht)
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| O2, NADPH/H+
H2O, NADP+ |
Häm-Thiolat | Steroid-21-Monooxygenase (Steroid-21-Hydroxylase) | 1.14.99.10 | Ox | AGS III | |||||
|
||||||||||

| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| O2, Red. Akz.
H2O, Ox. Akz. |
Häm-Thiolat | Steroid-21-Monooxygenase (Steroid-21-Hydroxylase) | 1.14.99.10 | Ox | AGS III | |||||
|
||||||||||

| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| O2, Red. Akz.
H2O, Ox. Akz. |
Häm-Thiolat | Steroid-21-Monooxygenase (Steroid-21-Hydroxylase) | 1.14.99.10 | Ox | AGS III | |||||
|
||||||||||
5. Spalte (senkrecht)
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| NAD(P)+ | 3β-Hydroxy-δ5-Steroid- Dehydrogenase | 1.1.1.145 | Ox | AGS II | ||||||
| Steroid-δ-Isomerase | 5.3.3.1 | Iso | ||||||||
|
||||||||||
| O2, Red. Ferredoxin
H2O, Ox. Ferredoxin |
Häm- Thiolat | Steroid-11β-Monooxygenase (Steroid-11β-Hydroxylase) | 1.14.15.4 | Ox | AGS IV | |||||
|
||||||||||
6. Spalte (waagerecht)
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| NADP+
NADPH/H+ |
11β-OH-Steroid-Dehydrogenase | 1.1.1.146 | Ox | Apparent mineralocorticoid excess (AME) Typ I, Typ II | ||||||
|
||||||||||

Biosynthese der Androgene und Östrogene
BearbeitenPregnenolon ⇒ 17α-OH-Pregnenolon ⇒ Dehydroepiandro- ⇒ Androstendiol
1.14.99.9 4.1.2.30 steron (DHEA) 1.1.1.51
⇓ ⇑ 1.1.1.145/ ⇓ ⇑ 1.1.1.145/
5.3.3.1 5.3.3.1 ⇓ 1.1.1.145/ ⇓ 1.1.1.145/
5.3.3.1 5.3.3.1
Progesteron ⇒ 17α-OH-Progesteron ⇒ Androstendion ⇒ Testosteron ⇒ 5α-DHT
1.14.99.9 4.1.2.30 1.1.1.51/ 1.3.99.5
⇓ 1.14.15.4 ⇓ 1.14.15.4 ⇓ 1.14.14.1 1.1.1.62 ⇓ 1.14.14.1
⇓ ⇓
11β-OH-Progesteron ⇒ 21-Deoxycortisol
1.14.99.9 Estron ⇔ Estradiol
1.1.1.51/
1.1.1.62
Reaktionen
4. Spalte (waagerecht)
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| O2, NADPH/H+
Acetat, H2O, NADP+ |
17-α-Hydroxyprogesteron-Aldolase | 4.1.2.30 | Ly | |||||||
|
||||||||||
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| 17-α-Hydroxyprogesteron-Aldolase | 4.1.2.30 | Ly | ||||||||
|
||||||||||

5. Spalte (senkrecht)
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| NAD(P)+ | NAD(P)+ | 3β-Hydroxy-δ5-Steroid- Dehydrogenase | 1.1.1.145 | Ox | AGS II | |||||
| Steroid-δ-Isomerase | 5.3.3.1 | Iso | ||||||||
|
||||||||||
| O2, NADPH/H+
H2O, NADP+ |
Häm-Thiolat | Unspez. Monooxygenase (CYP19A1, Aromatase) | 1.14.14.1 | Ox | Aromatase-Def. | |||||
|
||||||||||
| O2, NADPH/H+
2 H2O, NADP+ |
? | |||||||||
|
||||||||||
| O2, NADPH/H+
Formiat, H2O, NADP+ |
? | |||||||||
|
||||||||||



6. Spalte (waagerecht)
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| NAD(P)H/H+
NAD(P)+ |
3(oder 17)β-OH-Steroid-Dehydrogenase (?) | 1.1.1.51 | Ox | |||||||
|
||||||||||

| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| NAD(P)H/H+
NAD(P)+ |
3(oder 17)β-OH-Steroid-Dehydrogenase (?) | 1.1.1.51 | Ox | |||||||
|
1.1.1.62 | Ox | Männl. Pseudoherma- phroditismus | |||||||
|
||||||||||
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| NAD(P)H/H+
NAD(P)+ |
3(oder 17)β-OH-Steroid- Dehydrogenase (?) | 1.1.1.51 | Ox | |||||||
|
1.1.1.62 | Ox | Männl. Pseudoherma- phroditismus | |||||||
|
||||||||||
7. Spalte (senkrecht)
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| NAD(P)+ | NAD(P)+ | 3β-Hydroxy-δ5-Steroid- Dehydrogenase | 1.1.1.145 | Ox | AGS II | |||||
| Steroid-δ-Isomerase | 5.3.3.1 | Iso | ||||||||
|
||||||||||
| O2, NADPH/H+
H2O, NADP+ |
Häm-Thiolat | Unspez. Monooxygenase (CYP19A1, Aromatase) | 1.14.14.1 | Ox | Aromatase-Def. | |||||
|
||||||||||
| O2, NADPH/H+ (?)
2 H2O, NADP+ (?) |
? | |||||||||
|
||||||||||
| O2, NADPH/H+ (?)
Formiat, H2O, NADP+ (?) |
? | |||||||||
|
||||||||||


8. Spalte (waagerecht)
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| Red. Akz.
Ox. Akz. |
Steroid-5α-Reduktase | 1.3.99.5 | Ox | Männl. Pseudo- hermaphroditismus | ||||||
|
||||||||||

Enzyme
Bearbeiten| Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|
| Häm-Thiolat | Cholesterol-Monooxygenase (side-chain-cleaving) Cholesterol-20-22-Desmolase | 1.14.15.6 | Ox | (AGS I durch STAR-Def.) |
| 3β-OH-δ5-Steroid-Dehydrogenase (3β-OH-Steroid-Dehydrogenase) | 1.1.1.145 | Ox | AGS II | |
| Steroid-δ-Isomerase | 5.3.3.1 | Iso | ||
| Häm-Thiolat | Steroid-11β-Monooxygenase (Steroid-11β-Hydroxylase) | 1.14.15.4 | Ox | AGS IV |
| Häm-Thiolat | Steroid-21-Monooxygenase (Steroid-21-Hydroxylase) | 1.14.99.10 | Ox | AGS III |
| Häm-Thiolat | Corticosteron-18-Monooxygenase (Corticosteron-18-Hydroxylase) | 1.14.15.5 | Ox | Aldosteron-Def. I, Typ II |
| Häm-Thiolat | Steroid-17α-Monooxygenase (Steroid-17α-Hydroxylase) | 1.14.99.9 | Ox | AGS V |
| 11β-OH-Steroid-Dehydrogenase | 1.1.1.146 | Ox | Apparent mineralocorticoid excess (AME) Typ I, Typ II | |
| 17α-OH-Progesteron-Aldolase | 4.1.2.30 | Ly | ||
| 3(oder 17)β-OH-Steroid-Dehydrogenase | 1.1.1.51 | Ox | ||
| Häm-Thiolat | Unspez. Monooxygenase | 1.14.14.1 | Ox | |
| Estradiol-17β-Dehydrogenase (17β-OH-Steroid-Dehydrogenase) | 1.1.1.62 | Ox | ||
| Steroid-5α-Reduktase | 1.3.99.5 | Ox | Männl. Pseudohermaphroditismus |
Eigenschaften der Hormone
Bearbeiten |
 |
 |
 |
 |
 |
Progesteron
Bildungsorte: Corpus luteum, Plazenta
Stimulatoren: LH
Biologische Funktionen: Progesteron (synthetisch: Gestagen) ist das dominierende Hormon der 2. Zyklushälfte und das schwangerschaftserhaltende Hormon. Hemmung der Estradiolrezeptoren -> Hemmung der LH-Sekretion -> Hemmung der Ovulation. Erhöhung der Viskosität des Zervikalschleims. Erhöhung der Körpertemperatur. Antimineralkortikoid. katabol.
Mineralokortikoide (Aldosteron)
Bildungsorte: Aldosteron wird in der Zona glomerulosa der Nebennierenrinde gebildet.
Stimulatoren: Renin-Angiotensin-Aldosteron-System (RAAS)
Biologische Funktionen: Aldosteron fördert die Natrium- und Wasserrückresorption (Zunahme des EZV) durch vermehrte Biosynthese von Natriumkanalproteinen, die bes. im spätdistalen Tubulus der Niere in die luminale Membran eingebaut werden. Sekundär erhöhte Kaliumausscheidung.
Pathobiochemie:
- Hyperaldosteronismus (CONN-Syndrom): Natriumretention, Hypertension, Ödeme, Hypokaliämie
- Hypoaldosteronismus: Salzverlust (d.h. Natriumverlust), Exsikkose, Hypotension, Hyperkaliämie.
- Nebennierenrindeninsuffizienz (Morbus ADDISON). Ät.: primär (NNR-Schädigung z.B. autoimmunologisch, Tumor), sekundär (Hypophyseninsuffizienz). Pg.: Cortisol- und Aldosteronmangel. Klinik: Bräunliche Hyperpigmentierung, Schwächegefühl, Ermüdbarkeit, Hypotonie, Libidoverlust, gastrointestinale Symptome wie Bauchschmerzen, Obstipation, Diarrhoe. Kompl.: In Stresssituationen lebensbedrohliche ADDISON-Krise mit Koma, Blutdruckabfall, Dehydratation, Hypoglykämie, Pseudoperitonitis.
Glukokortikoide (Cortisol, Cortison u.a.)
Bildungsorte: Zona fasciculata der Nebennierenrinde
Stimulatoren: ACTH
Biologische Funktionen: Glukokortikoide wirken entzündungshemmend und immunsuppressiv. Mineralkortikoide Restwirkung! Katabol: Proteinsynthese↓, Proteolyse↑, Glykogenolyse↑, Gluconeogenese↑, Glykolyse↓, Lipolyse↑. Hemmung der Kalziumaufnahme im Darm, Kalzium-Mobilisation aus dem Knochen, vermehrte Kalziumausscheidung über die Niere -> Osteoporose.
Pathobiochemie:
- Hypercortisolismus (CUSHING-Syndrom): Klinik: Stammfettsucht, Vollmondgesicht, Striae distensae, Hypertonie, Hypokaliämie, diabetogene Stoffwechsellage, Fettleber, Osteoporose, Infektneigung (opportunistische Infektionen), Hautatrophie.
- Hypocortisolismus, Kortikoidentzugssyndrom
- Nebennierenrindeninsuffizienz (s.o.)
Androgene (Testosteron, Androsteron, Androstendion, Dehydroepiandrosteron (DHEA), Dihydrotestosteron (DHT))
Bildungsorte: Zona reticularis der Nebennierenrinde, Testes (LEYDIG-Zellen)
Stimulatoren: LH
Biologische Funktionen: Embryonale männliche Differenzierung des äußeren (DHT) und inneren (Testosteron) Genitale, Virilisierung, verstärkte Talgproduktion, verstärkte Blutbildung (Testosteron), Alopezie (DHT), anabol, Prostata-Wachstum (DHT).
Östrogene (Estron, Estradiol u.a.)
Bildungsorte: Ovar (reifender Follikel), Fettgewebe
Stimulatoren: FSH
Biologische Funktionen: Ausbildung der primären und sekundären weiblichen Geschlechtsmerkmale. Östrogen ist das dominierende Hormon der 1. Zyklushälfte. Es senkt die Viskosität des Zervikalschleims (erhöhte Spinnbarkeit, Farnkrautphänomen), hat eine mineralkortikoide Restwirkung, fördert die hepatische Proteinbiosynthese (HDL, Gerinnungsfaktoren, Hormontransportproteine), hemmt den Knochenabbau und fördert die Pigmentierung der Haut.
Pathobiochemie der Enzymdefekte
Bearbeiten| Erkrankung | Enzymdefekt | Epidemiologie | Folgen/Klinik |
|---|---|---|---|
| AGS I | STAR (20-22-Desmolase) | Aut.-rez.. 20,22-Desmolase-Def., schwerste Form des AGS: Androgenmangel -> m: Feminisierung. Aldosteronmangel -> Salzverlust-Syndrom. | |
| AGS II | 3β-Hydroxysteroid- Dehydrogenase | sehr selten | Störung der Biosynthese von Aldosteron, Kortisol und Sexualsteroiden. w: keine Virilisierung, m: Hypospadie, evtl. männl. Pseudohermaphroditismus. Aldosteronmangel -> Salzverlust. Labor: DHEA und DHEA-Sulfat erhöht, bei normalem Testosteron- und Androstendion-Spiegel. Erhöhtes 17α-Hydroxypregnenolon (evtl. erst nach ACTH-Test), erhöhter 17-OH-Pregnenolon/ 17-OH-Progesteron-Quotient (evtl. erst nach ACTH-Test). |
| AGS III | 21-Hydroxylase | Häufigste Form (95 %), ~ 1:10.000 (Heterozygoten- frequenz 1:40) | Aut.-rez.. Störung der Aldosteron- und Kortisolbiosynthese mit ACTH-Anstieg und Akkumulation der Vorstufen -> Androgenexzess -> Virilisierung, Hodentumoren beim Erwachsenen, Kleinwuchs durch vorzeitigen Schluss der Epiphysenfugen. Aldosteronmangel -> Salzverlust. Hypertension. Gynäkomastie beim Erwachsenen. Labor: Im Blut sind 17α-Hydroxyprogesteron und 21-Desoxycortisol erhöht (evtl. erst nach ACTH-Test). Im Harn ist Pregnantriol, eine Metabolit des 17α-Hydroxyprogesteron, erhöht. |
| AGS IV | 11β-Hydroxylase | ~ 1:100.000 | Störung der Aldosteron- und Kortisolbiosynthese mit ACTH-Anstieg und Akkumulation der Vorstufen -> Androgenexzess -> Virilisierung. Akkumulation des mineralkortikoid wirkenden 11-Deoxycorticosteron -> Hochdruck. Labor: 11-Desoxycorticosteron und 11-Desoxycortisol sind erhöht (evtl. erst nach ACTH-Test). 17α-Hydroxyprogesteron ist ebenfalls erhöht (DD: 21-Hydroxylase-Mangel). Die Metabolite der erstgenannten, Tetrahydro-Desoxycorticosteron und Tetrahydro-11-Desoxycortisol, sind im Harn erhöht. |
| AGS V | 17α-Hydroxylase | Aut.-rez.. Die Biosynthese von Glukokortiko- und Sexualsteroiden ist gestört -> erhöhtes ACTH, FSH und LH. Hohe Corticosteron- und Deoxycorticosteron-Spiegel -> Hypertension, hypokaliämische Alkalose. Aldosteron kann erhöht oder vermindert sein. Fehlende Sexualsteroidbiosynthese -> Primäre Amenorrhoe, männl. Pseudohermaphroditismus. Gynäkomastie. Labor: ACTH und FSH erhöht. | |
| Aldosteron-Def. I | 18-Hydroxylase | Aut.-rez.. Hypoaldosteronismus (Hypotension, Hyponatriämie, Hyperkaliämie). | |
| Aldosteron-Def. II | 11,18-Hydroxylase | Aut.-rez., Hypoaldosteronismus | |
| Apparent mineralocorticoid excess (AME) I | 11β-Hydroxysteroid- Dehydrogenase | Pseudohyperaldosteronismus (Hypertension, Hypokaliämie, aber niedrige Renin- und Aldosteronspiegel) durch Akkumulation von Kortisonvorstufen mit mineralokortikoider Restwirkung (?). | |
| Apparent mineralocorticoid excess (AME) II | 11β-Hydroxysteroid- Dehydrogenase | Pseudohyperaldosteronismus | |
| Männl. Pseudo- hermaphroditismus | Testosteron- 17β- Dehydrogenase | Aut.-rez.. Störung der Testosteronbiosynthese aus Androstenedion in den fetalen Testes -> Männl. Pseudohermaphroditismus (weibl. aussehende Genitalien bei Geburt), Leistenhoden. Normale Virilisierung in der Pubertät (sekundäre Restitution der Testosteronbiosynthese). Gynäkomastie, Infertilität, Schilddrüsenunterfunktion. | |
| Männl. Pseudo- hermaphroditismus (Pseudovaginale perineoskrotale Hypospadie) | 5α-Reduktase | Der Enzymdefekt führt zu einem Mangel an 5α-Dihydrotestosteron (5α-DHT). 5α-DHT ist für die embryonale Differenzierung des Urogenitalsinus zum äußeren männlichen Genitale notwendig. In der Folge kommt es zu einer schweren skrotalen oder perinealen Hypospadie, pseudovaginalen Einbuchtung und Kryptorchismus. |
Beim Adrenogenitalen Syndrom (AGS, (congenitale) adrenale Hyperplasie (CAH)) werden klinisch 4 Formen unterschieden: salt-wasting (SW), simple virilizing (SV), nonclassic (NC) late-onset (also called attenuated and acquired), and cryptic. Die Symptome richten sich neben dem betroffenen Enzym nach dem Ausmaß der Enzymdefizienz (verschiedene Allele) und der Gegenregulation des Organismus. Die Stimulation der Nebennierenrinde durch - je nach Enzymdefekt - erhöhte Hypophysenhormon- (ACTH, FSH, LH) oder Renin-Spegel führt zur Hyperplasie. Die vermehrt gebildeten Vorstufen fließen vermehrt in die funktionierenden Wege. Ein Salzverlust-Syndrom ist lebensbedrohlich. In Stresssituationen (Operationen, Infektionen) droht eine lebensbedrohliche ADISSON-Krise.
Die häufigste Form ist der 21-Hydroxylase-Mangel (AGS III). Hier kommt es zu einem Mangel an Aldosteron und Kortisol mit einem Überschuss an Sexualsteroiden.
Pharmakologie
BearbeitenDie Beeinflussung von Steroidhormonrezeptoren und Steroidhormon-metabolisierenden Enzymen spielt eine große Rolle in der Pharmakologie.
Steroidhormone werden z.B. eingesetzt zur Substitution (Bsp.: Hydrokortison bei Nebenniereninsuffizienz), zur Immunsuppression (Glucokortikoide bei allergischen Reaktionen, Autoimmunerkrankungen und zur Verhinderung von Abstoßungsreaktionen in der Transplantationsmedizin) und zur Kontrazeption (Östrogene und Gestagene in der „Pille“).
Das Wachstum Steroidhormon-abhängiger Tumoren lässt sich durch Rezeptorantagonisten (Bsp.: Tamoxifen bei Rezeptor-positivem Mammakarzinom) und Steroidhormonsynthesehemmer (Bsp.: Aromatase-Hemmer bei Rezeptor-positivem Mammakarzinom und Steroid-17α-Hydroxylase-Hemmer beim Prostatakarzinom) hemmen. 5α-Reduktase-Hemmer werden bei androgenetischem Haarausfall, bei benigner Prostatahyperplasie und Prostatakarzinom eingesetzt.
 |
 |
 |
Weblinks
Bearbeiten- KEGG: C21-Steroid hormone metabolism - Homo sapiens (human)
- KEGG: Androgen and estrogen metabolism - Homo sapiens (human)
- Laborlexikon - AGS
- med4you - Adrenogenitales-Syndrom (AGS)
Nukleotid-Stoffwechsel
BearbeitenEigenschaften und biologische Bedeutung
Bearbeiten

Nukleoside (nucleus (lat.): Kern) bestehen aus einer Purinbase (Adenin, Guanin) oder Pyrimidinbase (Cytosin, Thymin, Uracil) und einer Pentose (Ribose oder 2-Desoxyribose). Nukleotide besitzen zusätzlich ein bis drei Phosphatreste und sind die Bausteine der Desoxy- (DNA) und Ribonukleinsäuren (RNA). Die stickstoffhaltigen Basen bzw. Nukleotide werden nach ihrer Struktur und ihrem unterschiedlichen Synthese- und Abbauweg eingeteilt in Purine (heterozyklische Doppelringe, ein Imidazol-Ring und ein Pyrimidin-Ring) und Pyrimidine (heterozyklische Einfachringe). In der DNA liegen sich aus sterischen Gründen immer ein Purin und ein Pyrimidin gegenüber, genauer: Adenin paart mit Thymin und Guanin mit Cytosin. Die Basenpaarung wird durch Wasserstoffbrückenbindungen stabilisiert. Während die DNA aus Desoxyribonukleotiden aufgebaut ist und meist als Doppelhelix vorliegt besteht die RNA aus Ribonukleotiden, die Base Thymin ist durch Uracil ersetzt und sie liegt auch häufiger einzelsträngig vor als die DNA. Für die Basenpaarung müssen Guanin, Cytosin und Thymin in ihrer Keto-Form (Lactam-Form) vorliegen.
Weiterhin dienen insbesondere die Purintriphosphate ATP und GTP in der Zelle als mobile kleinmolekulare „Energielieferanten“ von chemischen Reaktionen, indem sie durch die Hydrolyse ihrer energiereichen Phosphorsäureanhydridbindung das Reaktionsgleichgewicht auf die Seite der Hydrolyse verschieben. ATP wird v.a. in den Mitochondrien aus ADP und anorganischem Phosphat von der ATP-Synthase regeneriert, aber auch durch Substratkettenphosphorylierung z.B. in der Glycolyse gewonnen. ATP treibt insbesondere biochemische Reaktionen des Intemediärstoffwechsels an, wie auch Proteinfunktionen, z.B. ATP-abhängige Pumpen wie die Natrium-Kalium-Pumpe. Weiterhin liefert ATP auch meist das Phosphat für die Phosphorylierung von Proteinen durch Proteinkinasen. GTP treibt v.a. molekularbiologische Prozesse an wie z.B. den Transport von Kernproteinen in den Zellkern (Importine), die Proteinbiosynthese am Ribosom und den Aufbau der Mikrotubuli.
GTP ist auch der Rohstoff der Biopterin-Biosynthese.
Zyklisches AMP (cAMP) und GMP (cGMP) dienen in vielen intrazellulären Signalkaskaden als wichtige sekundäre Botenstoffe.
Nukleotide dienen auch zur Aktivierung vieler Moleküle, insbesondere von Monosacchariden für die Erzeugung glycosidischer Bindungen. Solche aktivierten Moleküle sind z.B. GDP-Mannose, GDP-Fucose, UDP-Glucose, UDP-Galactose, UDP-Glucuronsäure, die Aminozucker UDP-N-Acetyl-Glucosamin, UDP-N-Acetyl-Galactosamin, UDP-N-Acetyl-Mannosamin und CMP-N-Acetylneuraminsäure, sowie das CDP-Cholin.
Der Abbau der Purine liefert Harnsäure, die renal durch tubuläre Sekretion ausgeschieden wird.
Nomenklatur
Bearbeiten| Base | Ribonukleosid | Ribonukleotid | Desoxyribonukleotid |
|---|---|---|---|
| Adenin | Adenosin | Adenosin-mono/di/tri-phosphat (AMP, ADP, ATP) | dAMP, dADP, dATP |
| Guanin | Guanosin | Guanosin-mono/di/tri-phosphat (GMP, GDP, GTP) | dGMP, dGDP, dGTP |
| Thymin | Thymidin | Thymidin-mono/di/tri-phosphat (TMP, TDP, TTP) | dTMP, dTDP, dTTP |
| Cytosin | Cytidin | Cytidin-mono/di/tri-phosphat (CMP, CDP, CTP) | dCMP, dCDP, dCTP |
| Uracil | Uridin | Uridin-mono/di/tri-phosphat (UMP, UDP, UTP) | dUMP, dUDP, dUTP |
Desoxyribonukleosiden/-tiden wird ein „Desoxy-“ vorangestellt, der jeweiligen Abkürzung entsprechend ein kleines d.
Purin-Stoffwechsel
BearbeitenAllgemeines
BearbeitenPurine sind wichtige Biomoleküle. Sie bilden neben den Pyrimidinen die Bausteine der DNA, dienen als mobile Energiewährung und sind Ausgangsstoff des Biopterins und wichtiger intrazellulärer Signalmoleküle. Das primäre Abbauprodukt der Purine ist die Harnsäure.
Biosynthese von Inosin-5'-monophosphat (IMP)
Bearbeiten| All. | ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||
| + Pi
- ADP, GDP |
ATP
AMP |
Ribose-phosphat- Diphosphokinase | 2.7.6.1 | Tr | PRPS1-assoziierte Syndrome | ||||||
|
|||||||||||
| + PRPP
- AMP, ADP, ATP, GMP, GDP, GTP |
Glutamin
PPi, Glutamat |
Amidophosphoribosyl- transferase | 2.4.2.14 | Tr | |||||||
|
|||||||||||
| ATP, Glycin
ADP + Pi |
Phosphoribosylamin--Glycin-Ligase | 6.3.4.13 | Lig | ||||||||
|
|||||||||||
| N10-Formyl-THF | Phosphoribosylglycinamid- Formyltransferase | 2.1.2.2 | Tr | ||||||||
|
|||||||||||
| ATP, Glutamin
ADP + Pi, Glutamat |
Phosphoribosylformyl- glycinamidin-Synthase | 6.3.5.3 | Lig | ||||||||
|
|||||||||||
| ATP
H2O, ADP + Pi |
Phosphoribosylformyl- glycinamidin-Cyclo-Ligase | 6.3.3.1 | Lig | ||||||||
|
|||||||||||
| CO2
|
Phosphoribosylaminoimidazol- Carboxylase | 4.1.1.21 | Ly | ||||||||
|
|||||||||||
| ATP, Aspartat
ADP + Pi |
Phosphoribosylaminoimidazol- succinocarboxamid-Synthase | 6.3.2.6 | Lig | ||||||||
|
|||||||||||
| Adenylosuccinat-Lyase | 4.3.2.2 | Ly | ADSL-Def. (Succinyl-purinämischer Autismus) | ||||||||
|
|||||||||||
| N10-Formyl-THF | Phosphoribosylaminoimidazol- carboxamid-Formyltransferase | 2.1.2.3 | Tr | AICA-Ribosurie | |||||||
|
|||||||||||
|
|
IMP-Cyclohydrolase | 3.5.4.10 | Hyd | AICA-Ribosurie | |||||||
|
|||||||||||











 |
 |
Allgemeines
BearbeitenDer fertige Purinkern besteht aus 5 Kohlenstoff- und 4 Stickstoffatomen. Der Kohlenstoff stammt aus Glycin (2 C), 2 Formylresten aus 2 N10-Formyl-Tetrahydrofolsäure und 1 Kohlendioxid. Der Stickstoff stammt aus den nicht-essentiellen Aminosäuren Glycin, Glutamin (2x) und Aspartat.
Chemisch betrachtet besteht das Molekül aus zwei verschiedenen zusammenhängenden stickstoffhaltigen Ringsystemen (ein Pyrimidin und ein Imidazol), mit aromatischen Eigenschaften.
Die Purinbasen sind über ihr N-Atom 9 mit dem C-Atom 1' der Ribose verbunden.
Die Reaktionen im Detail
BearbeitenDas Trägermolekül Ribose-5-phosphat, an dem die Purinbase synthetisiert wird, stammt aus dem Pentosephosphatweg. Zuvor muss es ATP-abhängig zu Phosphoribosylpyrophosphat (PRPP) phosphoryliert und damit „aktiviert“ werden. Da PRPP auch für die Pyrimidin- und NAD-Biosynthese sowie für die Purin-Wiederverwertung verwendet wird folgt der Beginn der eigentlichen de novo Purin-Synthese und ihre Regulation erst mit dem nächsten Schritt.
Ausgehend vom PRPP wird der Purinrest Inosin am C1-Atom des Phophoriboserestes in 10 Reaktionsschritten aufgebaut mit dem Nukleotid Inosin-5'-monophosphat (IMP) als (vorläufiges) Endprodukt. Was passiert nun im Einzelnen?
Im 1. Schritt erfolgt die Abspaltung von Pyrophosphat. Dies liefert genug Energie, um die Aminogruppe von Glutamin auf den Phosphoriboserest zu übertragen, so dass 5-Phosphoribosylamin (PRA) entsteht. Katalysiert wird die Reaktion von der Amidophosphoribosyltransferase. Das Enzym reguliert als Gatekeeper den Substratdurchfluss durch diesen Stoffwechselweg und wird entsprechend vom Feedforward-Aktivator PRPP allosterisch aktiviert und von den Endprodukten des Syntheseweges AMP, ADP, ATP, GMP, GDP und GTP allosterisch inhibiert (Feedback-Hemmung).
Im 2. Schritt katalysiert die Phosphoribosylamin--Glycin-Ligase die Anlagerung von Glycin an die Aminogruppe des 5-Phosphoribosylamins, womit das zweite von vier Stickstoffatomen eingebaut wäre und nun Glycinamidribonukleotid (GAR) vorliegt.
Im 3. Schritt wird ein Formylrest, der von N10-Formyl-Tetrahydrofolsäure angeliefert wird, an das Glycinamidribonukleotid angelagert, katalysiert von der Phosphoribosylglycinamid-Formyltransferase. Das Reaktionsprodukt ist Formylglycinamidribonukleotid (FGAR).
Im 4. Schritt liefert Glutamin ein weiteres Stickstoff-Atom, das unter ATP-Verbrauch von der Phosphoribosylformylglycinamidin-Synthase eingebaut wird und damit Formylglycinamidinribonukleotid (FGAM) liefert.
Im 5. Schritt erfolgt unter dem Einfluss der Phosphoribosylformylglycinamidin-Cyclo-Ligase der Ringschluß des Imidazol-Rings unter Wasserabspaltung, dabei kommt 5-Aminoimidazolribonukleotid (AIR) heraus. Die Reaktion kostet ein weiteres ATP.
Im 6. Schritt wird 5-Aminoimidazolribonukleotid (AIR) am C5-Atom des Imidazolrestes von der Phosphoribosylaminoimidazol-Carboxylase zum 5-Aminoimidazol-4-carboxylatribonukleotid (CAIR) carboxyliert.
Der 7. und 8. Reaktionsschritt sorgt nun via Aspartat-Zyklus für den Einbau des vierten Stickstoffatoms in den Purinkern. Auch das kostet wieder ein ATP. Die Kondesation mit Aspartat wird von der Phosphoribosylaminoimidazolsuccinocarboxamid-Synthase vermittelt, die folgende Abspaltung von Fumarat von der Adenylosuccinat-Lyase.
Der 9. Schritt erfordert einen weiteren Formylrest, der wieder von N10-Formyl-Tetrahydrofolsäure geliefert wird. Die Abhängigkeit vom Folsäure-Angebot erklärt zwanglos, warum ein Folat-Mangel die de novo Purinbiosynthese und damit die DNA-Synthese und Zellteilung beeinträchtigen kann. Die Phosphoribosylaminoimidazolcarboxamid-Formyltransferase katalysiert die Reaktion.
Im 10. Schritt wird auch der zweite Ring - der Pyrimidin-Ring - unter Wasserabspaltung von der IMP-Cyclohydrolase geschlossen. Der Purin-Kern ist nun fertig und kann weiter modifiziert werden.
Pharmakologie
BearbeitenDie Purinbiosynthese kann durch Blockade des Folat-Stoffwechsels behindert werden, z.B. mit dem Dihydrofolatreduktase-Hemmer Methotrexat (MTX). In der Folge kommt es zu einem Mangel an DNA-Bausteinen und zu einer Hemmung der Zellvermehrung vor allem von proliferationsfreudigen Geweben. Dies macht man sich in der Therapie von Krebs (Tumorzellen) und Autoimmunerkrankungen (Immunzellen) zunutze.
Synthese von AMP aus IMP und Desaminierung zu IMP
Bearbeiten| All. | ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||
| + GTP | GTP, Aspartat
GDP + Pi |
Adenylosuccinat-Synthase | 6.3.4.4 | Lig | |||||||
|
|||||||||||
| Adenylosuccinat-Lyase | 4.3.2.2 | Ly | ADSL-Def. (Succinyl-purinämischer Autismus) | ||||||||
|
|||||||||||
| H2O
NH3 |
AMP-Deaminase | 3.5.4.6 | Hyd | MADA-Defizienz, AMPD3-Defizienz | |||||||
|
|||||||||||

Die vorgenannten Reaktionen dienen nicht nur der Biosynthese von AMP im Rahmen der de novo Purinbiosynthese. Als sogenannter Purinnukleotidzyklus spielen sie eine wichtige Rolle im Skelettmuskel unter Belastung. Hier dient die Reaktionsfolge der Auffüllung des Citratzyklus mit Fumarat, das zuvor als Aspartat via Proteolyse in der Muskelzelle freigesetzt wurde (im Gegensatz zur reinen AMP-Synthese würde die Aspartat-Rekrutierung aus Oxalacetat, ebenfalls ein Citratzyklus-Intermediat, im Aspartatzyklus keinen Sinn machen). Ein weiterer Punkt ist der AMP-Spiegel, der durch die Deaminase-Reaktion konstant gehalten wird. Die Freisetzung von Ammoniak lässt sich laborchemisch als Ammoniakanstieg messen. Ein Defekt dieses Systems auf Ebene der AMP-Deaminase (MADA-Defizienz) ist die am weitesten verbreitete Ursache einer Myopathie.
Synthese von GMP aus IMP und Desaminierung zu IMP
Bearbeiten| All. | ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||
| H2O, NAD+ | IMP-Dehydrogenase | 1.1.1.205 | Ox | Retinitis pigmentosa 10 / Leber'sche kongenitale Amaurose 11 | |||||||
|
|||||||||||
| + ATP | H2O, ATP, Glutamin
AMP + PPi, Glutamat |
GMP-Synthase (Glutamin-hydrolysierend) | 6.3.5.2 | Lig | |||||||
|
|||||||||||
| NADPH/H+
NH3, NADP+ |
GMP-Reduktase | 1.7.1.7 | Ox | ||||||||
|
|||||||||||
Pharmakologie: Mycophenolat mofetil (MMF, MPA) hemmt die IMP-Dehydrogenase (IMPDH) und damit die GMP-Synthese. Dies beeinträchtigt wiederum die DNA-Biosynthese und Zellteilung vor allem von Lymphozyten, die ihren Purinbedarf vorwiegend über die de novo Purinbiosynthese decken. Der Wirsktoff kann daher als Immunsuppressivum z.B. zur Verhinderung einer Abstoßung nach Transplantation genutzt werden.
Regulation
BearbeitenDas Schrittmacherenzym der Purin-Biosynthese, die Amidophosphoribosyltransferase wird durch die Endprodukte der Purin-Bildung gehemmt und durch PRPP stimuliert.
Nach der Verzweigungsstelle IMP findet man einen sinnigen Mechanismus, der beide Zweige im Gleichgewicht hält. ATP stimuliert die GMP-Synthese, so dass das IMP eher dort hin fließt. GTP stimuliert wiederum die AMP-Synthese, so dass das IMP wieder vermehrt in die andere Richtung fließt.
Stoffwechsel der Adenin- und Guaninnucleotide
Bearbeiten
- Die Nukleotide AMP und GMP und ihre Deoxyderivate dAMP und dGMP stellen die eine Hälfte der Bausteine der Nukleinsäuren (RNA und DNA).
- ATP und GTP dienen der Zelle als universelle „Energiewährung“, die vielen biochemischen Reaktionen die Energie liefert (die ATP-Hydrolyse ist stark exergon und verschiebt das Reaktionsgleichgewicht auf die Seite der Hydrolyse). Das ATP wird hauptsächlich von der ATP-Synthase an der inneren Mitochondrienmembran unter Sauerstoffverbrauch aus ADP und Pi erzeugt (Oxidative Phosphorylierung), zum Teil auch in der Glycolyse gewonnen (Substratkettenphosphorylierung). Letzteres wird von der Zelle besonders unter anaeroben Bedingungen genutzt. Eine weitere Substanz, die ein so hohes Gruppenübertragungspotential hat, dass sie ihr Phosphat auf ADP übertragen kann ist das Kreatinphosphat. Da das Kreatinphosphat das Phosphat vorher vom ATP übernommen hat entsteht hier netto kein neues ATP, Kreatinphosphat fungiert hier nur als „Zwischenspeicher“, um kurzzeitige ATP-Versorgungsengpässe auszugleichen. GTP entsteht z.B. indem GDP ein Phosphat von ATP übernimmt oder durch Substratkettenphosphorylierung im Citratzyklus. ATP treibt viele biochemische Reaktionen des Intermediärstoffwechsels an wie auch Proteine, z.B. ATP-abhängige Pumpen wie die Natrium-Kalium-Pumpe. Weiterhin liefert ATP auch meist das Phosphat für die Phosphorylierung von Proteinen durch Proteinkinasen. GTP treibt v.a. molekularbiologische Prozesse an wie z.B. den Transport von Kernproteinen in den Zellkern (Importine), die Proteinbiosynthese am Ribosom und den Aufbau der Mikrotubuli.
- ATP und GTP können von G-Protein-gesteuerten Enzymen (Adenylatzyklase, Guanylatzyklase) in zyklisches AMP (3',5'-cyclo-AMP, cAMP) und GMP (cGMP) umgewandelt werden, die wichtige second messenger darstellen (Botenstoffe der intrazellulären Signalübertragung).
- GTP ist auch der Rohstoff der Biopterin-Biosynthese.
Im Folgenden sind beispielhaft die möglichen Reaktionen der Adeninnukleotide tabellarisch aufgeführt:
| Reaktion | Co. | Enzym | EC | EG | Erkr. | ||
|---|---|---|---|---|---|---|---|
| AMP + ATP | ⇔ | 2 ADP | Adenylat-Kinase | 2.7.4.3 | Tr | AK1-Def. (Hämolyt. Anämie) | |
|
ADP + H2O ATP + H2O ATP + H2O |
⇒
⇒ ⇒ |
AMP + Pi
AMP + 2 Pi ADP + Pi |
Ca | Apyrase | 3.6.1.5 | Hyd | |
| ADP + Pi | ⇔ | ATP + H2O | FoF1-ATPase | 3.6.3.14 | Hyd | MTATP6-Def. (Leigh-S., NARP-S., Leber Opticusatrophie) | |
| ATP + NDP | ⇔ | ADP + NTP | Nucleosid-diphosphat-Kinase | 2.7.4.6 | Tr | ||
| ADP + 1,3-BPG | ⇔ | ATP + 3-PG | Phosphoglycerat-Kinase | 2.7.2.3 | Tr | PGK1-Def. | |
| ADP + PEP | ⇒ | ATP + Pyruvat | Pyruvat-Kinase | 2.7.1.40 | Tr | PKLR-Def. | |
| ATP + Kreatin | ⇔ | ADP + Kreatin-P | Kreatin-Kinase | 2.7.3.2 | Tr | ||
| ATP + H2O | ⇒ | ADP + Pi | Adenosintriphosphatase | 3.6.1.3 | Hyd | Hämolyt. Anämie | |
| NTP + RNAn | ⇔ | PPi + RNAn+1 | DNA-abhängige RNA-Polymerase | 2.7.7.6 | Tr | ||
| RNAn+1 + Pi | ⇔ | RNAn + NDP | Polyribonucleotid-Nucleotidyltransferase | 2.7.7.8 | Tr | ||
| ATP | ⇒ | 3',5'-cyclo-AMP + PPi | Adenylat-Cyclase | 4.6.1.1 | Ly | ||
| 3',5'-cyclo-AMP + H2O | ⇒ | AMP | 3',5'-cyclo-Nucleotid-Phosphodiesterase | 3.1.4.17 | Hyd | ||
| ADP + Thioredoxin | ⇒ | dADP + Thioredoxin- Disulfid + H2O | Ribonucleosid-diphosphat-Reduktase | 1.17.4.1 | Ox | ||
| ATP + dNDP | ⇔ | ADP + dNTP | Nucleosid-diphosphat-Kinase | 2.7.4.6 | Tr | ||
| dADP + PEP | ⇒ | dATP + Pyruvat | Pyruvat-Kinase | 2.7.1.40 | Tr | PKLR-Def. | |
| dNTP + DNAn | ⇔ | PPi + DNAn+1 | DNA-abhängige DNA-Polymerase | 2.7.7.7 | Tr | ||
| dAMP + dATP | ⇔ | 2 dADP | Adenylat-Kinase | 2.7.4.3 | Tr | AK1-Def. (Hämolyt. Anämie) | |
| dAMP + H2O | ⇔ | Deoxyadenosin + Pi | 5'-Nucleotidase | 3.1.3.5 | Hyd | ||
| Deoxyadenosin + PO4H3 | ⇔ | Adenin + 2-Deoxy-D-Rib-1-P | Purinnucleosid-Phosphorylase | 2.4.2.1 | Tr | PNP-Defizienz | |
| Deoxyadenosin + H2O | ⇔ | Deoxyinosin + NH3 | Adenosin-Deaminase | 3.5.4.4 | Hyd | Severe combined immunodeficiency (SCID) | |
| Deoxyinosin + Pi | ⇒ | Hypoxanthin + 2-Deoxy-D-Rib-1-P | Purinnukleosid-Phosphorylase | 2.4.2.1 | Tr | PNP-Defizienz | |
Purine werden zu Harnsäure abgebaut
Bearbeiten| Alternative Darstellung (kompakt) |
| ⇓ | Subst. | (⇑) | ⇓ | Subst. | (⇑) | ⇓ | Subst. | (⇑) | ⇓ | Subst. | (⇑) |
|---|---|---|---|---|---|---|---|---|---|---|---|

AMP |

IMP |

XMP |

GMP | ||||||||
| H2O
Pi |
H2O
Pi |
H2O
Pi |
H2O
Pi |
||||||||
|
|
|||||||||||
| H2O
NH3 |
|||||||||||

Inosin |

Xanthosin |

Guanosin | |||||||||
| Pi | Pi | Pi | |||||||||

Hypoxanthin |

Guanin | ||||||||||
| O2, H2O
O2-, 2H+ |
H2O
NH3 |
||||||||||
 Xanthin Xanthin
| |||||||||||
| O2, H2O
O2-, 2H+ |
|||||||||||

Harnsäure (Ketoform) |

⇌ Harnsäure ⇌ (Enolform) |

Harnsäure (dissoz., pK = 5,4) | |||||||||

| Enzym | Co. | EC | EG | Erkr. |
|---|---|---|---|---|
| 1) 5'-Nucleotidase | 3.1.3.5 | Hyd | ||
| 2) Adenosin-Deaminase | 3.5.4.4 | Hyd | Severe combined immunodeficiency (SCID) | |
| 3) Purinnukleosid-Phosphorylase | 2.4.2.1 | Tr | PNP-Defizienz | |
| 4) Xanthin-Oxidase (XOD) | FAD, FeS, Mo | 1.17.3.2 | Ox | Xanthinurie I |
| 5) Guanin-Deaminase | 3.5.4.3 | Hyd |

Primaten, Vögel und einige Reptilien bauen Purine bis zur Harnsäure ab, die über den Urin ausgeschieden wird. Andere Tiere bauen die Harnsäure weiter ab zu besser wasserlöslichen Produkten wie Allantoin (die meisten Säugetiere), Harnstoff und Glyoxylsäure (Fische) oder Ammoniak (marine Invertebraten).
Harnsäure besitzt vermutlich eine antioxidative Wirkung, was den Verlust der Harnsäure-abbauenden Enzyme trotz des Nachteils der schlechteren Wasserlöslichkeit für Primaten u.a. Tiere lohnend gemacht haben könnte.
Pathobiochemie
BearbeitenEin übermäßiger Anfall von Harnsäure (purinreiche Kost (Innereien, Wurst), Tumorlyse) und/oder eine verschlechterte renale Ausscheidung (erbliche Transporter-Defekte, Alkohol (-> Acetat) und ASS durch Konkurrenz am tubulären Säuretransporter) führen zur Gicht. Hier kommt es durch die Überschreitung des Löslichkeitsprodukts zum Ausfällen von scharfkantigen Uratkristallen im Gewebe, die lokal eine Entzündungsreaktion hervorrufen.
Pharmakologie
BearbeitenDie Xanthinoxidase kann durch das Hypoxanthin-Analogon Allopurinol gehemmt werden. Die Harnsäurebildung nimmt dadurch ab, die Bildung der besser wasserlöslichen Metaboliten Xanthin und Hypoxanthin nimmt zu.
Salvage Pathway
Bearbeiten| All. | ( ⇓ ) | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||
| - AMP | PPi | Adenin-Phosphoribosyl- transferase (APRT) | 2.4.2.7 | Tr | APRT-Defizienz | ||||||
|
|||||||||||

| All. | ( ⇓ ) | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||
| - IMP, GMP | PPi | Hypoxanthin-Phosphoribosyl- transferase (HGPRT) | 2.4.2.8 | Tr | Kelley-Seegmiller-Syndrom, Lesch-Nyhan-Syndrom | ||||||
|
|||||||||||
| All. | ( ⇓ ) | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||
| - IMP, GMP | PPi | Hypoxanthin-Phosphoribosyl- transferase (HGPRT) | 2.4.2.8 | Tr | Kelley-Seegmiller-Syndrom, Lesch-Nyhan-Syndrom | ||||||
|
|||||||||||
| All. | ( ⇓ ) | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||
| - AMP | PPi | Adenin-Phosphoribosyl-transferase (APRT) | 2.4.2.7 | Tr | APRT-Defizienz | ||||||
| - IMP, GMP | Hypoxanthin-Guanin-Phosphoribosyl-transferase (HGPRT) | 2.4.2.8 | Tr | Kelley-Seegmiller-Syndrom, Lesch-Nyhan-Syndrom | |||||||
|
|||||||||||
Die Purinbasen Adenin, Hypoxanthin, Xanthin und Guanin können mit Hilfe zweier Enzyme wiederverwertet werden, indem sie mit einem Phosphoribosylrest wieder zum Nucleotid aufgebaut werden. Dies ist sehr sinnvoll, da damit erstens ATP eingespart werden kann (die Bildung eines Purins kostet mindestens 7 (AMP) bzw. 9 (GMP) energiereiche Phosphatbindungen) und zweitens die Harnsäurebildung drastisch reduziert wird.
Pathobiochemie
BearbeitenBeim X-chromosomal-rezessiven Lesch-Nyhan-Syndrom kommt es aufgrund eines Defektes der Hypoxanthin-Guanin-Phosphoribosyltransferase (HGPRT) zur fehlenden Rückgewinnung der Purinbasen und einem Überschuss an PRPP. Letzteres setzt zusammen mit dem Nukleotid-Mangel eine exzessive Purin- und damit Harnsäureproduktion in Gang. Die Erkrankung manifestiert sich in schweren neurologischen Störungen und Automutilation (Selbstverstümmelung), sowie Harnsäuresteinen. Eine abgeschwächte Form dieser Erkrankung mit einer HGPRT-Restfunktion stellt das Kelley-Seegmiller-Syndrom dar. Hier steht klinisch die Bildung von Harnsäuresteinen und die Harnsäurennephropathie im Vordergrund.
Weblinks
Bearbeiten- KEGG: Purine metabolism - Homo sapiens (human)
- RCSB PDB: Xanthine Oxidoreductase
- RCSB PDB: Hypoxanthine-guanine phosphoribosyltransferase (HGPRT)
Pyrimidin-Stoffwechsel
BearbeitenAllgemeines
BearbeitenPyrimidinnukleotide bilden neben den Purinnukleotiden die Bausteine der DNA und RNA. UTP und CTP dienen weiterhin als Aktivatoren verschiedener Biomoleküle.
Pyrimidine bestehen aus einem aromatischen, stickstoffhaltigen, sechsgliedrigen Heterozyklus.
Übersicht
Bearbeiten| Übersicht: Stoffwechsel der Pyrimidine | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Farbcode: Thioredoxin, Folat. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Die Pyrimidinbiosynthese beginnt mit der Synthese von Uridin-5'-monophosphat (UMP). Aus UMP werden dann die anderen Pyrimidinnukleotide gebildet, wie in der Übersicht zu sehen. Nach links muß Energie (meist ATP) investiert werden, nach rechts erfolgt der Abbau.
Wichtige Cofaktoren im Pyrimidinstoffwechsel sind Thioredoxin und Folat (5,10-Methylen-THF). Ersteres wird für die Bildung von Desoxyribonukleotiden benötigt (DNA!), letzteres liefert die Methylgruppe für die Bildung der Thymin-Base im 2'-Desoxythymidin-5'-monophosphat (dTMP).
Betrachtet man nur die Basen, so entsteht Cytosin formal aus Uracil, indem der Sauerstoff am C-Atom 4 durch eine Aminogruppe substituiert wird. Thymin entsteht aus Uracil durch Anheftung einer Methylgruppe an das C-Atom 5.

Cytosin |
⇐ | 
Uracil |
⇒ | 
Thymin |
 |
 |
Biosynthese von UMP
Bearbeiten| All. | ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||
| + PRPP
- UTP |
2 ATP, Glutamin
2 ADP, Pi, Glutamat |
Carbamoylphosphat-Synthase II (Glutamin) Zytosol | 6.3.5.5 | Lig | |||||||
|
|||||||||||
| Aspartat
Pi |
|
2.1.3.2 | Tr | ||||||||
|
|||||||||||
| - Orotat |
|
|
Zn | Dihydroorotase Zytosol |
3.5.2.3 | Hyd | |||||
|
|||||||||||
| - UMP | O2 | O2 | FAD, FMN | Dihydroorotat-Dehydrogenase
Mitochondrium |
1.3.3.1 | Ox | |||||
|
|||||||||||
| PRPP
PPi |
Orotat-Phosphoribosyltransferase
Zytosol |
2.4.2.10 | Tr | Orotacidurie I | |||||||
|
|||||||||||
| Orotidin-5'-phosphat- Decarboxylase
Zytosol |
4.1.1.23 | Ly | Orotacidurie I, Orotacidurie II | ||||||||
|
|||||||||||





Beteiligte Enzyme
BearbeitenDie sechs Reaktionsschritte der de novo Pyrimidin-Biosynthese sind evolutionär hochkonserviert. Bei Bakterien werden sie jeweils von einzelnen Enzymen katalysiert. Beim Säugetier werden die ersten drei und die letzten beiden Schritte jeweils von einem multifunktionellen Enzym katalysiert. Die Proteine untergliedern sich in folgende enzymatisch aktiven Bestandteile:
- CAD:
- Carbamoylphosphat-Synthetase (CPS II)
- Aspartat-Transcarbamoylase (ATCase)
- Dihydroorotase
- Dihydroorotat-Dehydrogenase (DHOdhase)
- UMP-Synthetase (UMPS):
- Orotat-Phosphoribosyltransferase (OPRT)
- Orotidin-5'-phosphat-Decarboxylase (ODC)
Die Dihydroorotat-Dehydrogenase ist im Bereich der inneren Mitochondrienmembran lokalisiert, die zwei anderen Proteine/Multienzymkomplexe finden sind im Zytosol.
Reaktionsschritte
BearbeitenDie ersten drei Schritte zur Bildung von Dihydroorotat werden vom CAD-Multienzymkomplex katalysiert.
Die zytosolische Carbamoylphosphat-Synthetase II bildet im 1. Schritt aus Bicarbonat, Glutamin und 2 ATP das Stickstoff- und Phosphathaltige Carbamoylphosphat und kontrolliert als Gatekeeper den Substratfluss durch diesen Biosyntheseweg. (Die Carbamoylphosphat-Synthetase I (CPS I) in den Lebermitochondrien produziert analog dazu Carbamoylphosphat für den Harnstoffzyklus.)
Die Aspartat-Carbamoyltransferase spaltet im 2. Schritt den Phosphatrest ab und nutzt die dabei frei werdende Energie, um die Aminosäure Aspartat anzulagern. Dabei entsteht Carbamoylaspartat. Aspartat bringt dabei das zweite Stickstoffatom und zwei Kohlenstoffatome mit für den zukünftigen Pyrimidin-Ring.
Die Dihydroorotase kondensiert nun im 3. Schritt die Carboxyl- und Aminogruppe unter Wasserabspaltung, wodurch das Ringsystem geschlossen wird. Dihydroorotat ist entstanden.
Im 4. Schritt oxidiert das in der inneren Mitochondrienmembran lokalisierte Flavoenzym Dihydroorotat-Dehydrogenase das Ringsystem, das dadurch eine zusätzliche dritte Doppelbindung erhält. Die Elektronen werden direkt über Ubichinon in die Atmungskette geschleust. Dabei entsteht Orotat.
Die letzten beiden Schritte werden von der UMP-Synthase katalysiert.
Die Orotat-Phosphoribosyltransferase (OPRT) verbindet im 5. Schritt Orotat mit dem Phosphoribosylrest von PRPP unter Abspaltung von Pyrophosphat zum Orotidin-5'-monophosphat (OMP).
Die Orotidin-5'-phosphat-Decarboxylase (ODC) decarboxyliert den Orotidin-Rest am C6-Atom, so das aus Orotidin-5'-monophosphat (OMP) Uridin-5'-monophosphat (UMP) wird.
Regulation
BearbeitenKurzfristig wird der Substratfluss bedarfsorientiert vor allem allosterisch auf Höhe der Carbamoylphosphat-Synthase II reguliert. Das Endprodukt UTP wird dabei als Feedback-Hemmer. PRPP wirkt als Feedforward-Aktivator, vermutlich um die Pyrimidinbiosynthese auf die ebenfalls PRPP-abhängige Purinbiosynthese abzustimmen.
Um die UMP-Synthese mittelfristig an die DNA-Synthese bzw. Zellproliferation (Zellteilung) anzupassen kann CAD an zwei verschiedenen Stellen phosphoryliert werden. Die Phosphorylierung von CAD durch die MAP-Kinase (nach Stimulation des zellteilungsfördernden EGF-Rezeptors) vor Beginn der S-Phase (DNA-Synthesephase) senkt den Einfluss des allosterischen Hemmers UTP und erhöht den Einfluss des allosterischen Aktivators PRPP, so dass die UMP-Synthese drastisch gesteigert wird. Die Phosphorylierung an einer anderen Domäne des CAD-Proteins durch die Proteinkinase A am Ende der S-Phase führt wieder den ursprünglichen Zustand herbei. So kann die Produktion von DNA-Bausteinen selektiv für die S-Phase gesteigert werden.
Weiterhin erfolgt eine Regulation auf Höhe der Genexpression. Der Transkriptionsfaktor C-Myc erhöht dabei die Transkription von CAD am G1/S-Übergang des Zellzyklus. Umgekehrt wird CAD im Rahmen der Apoptose unter dem Einfluss von Caspasen abgebaut.
Eine Fehlregulation mit dauerhaft aktivierter Pyrimidinbiosynthese bei unkontrollierter Zellteilung spielt in der Karzinogenese (Krebsentstehung) eine Rolle.
Stoffwechsel der Uridinphosphate
BearbeitenFreisetzung von UDP aus der RNA
Bearbeiten| All. | ⇓ | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||
| Pi
RNAn |
Polyribonukleotid- Nukleotidyltransferase | 2.7.7.8 | Tr | ||||||||
|
|||||||||||


Dephosphorylierung von UTP zu UMP
Bearbeiten| All. | ⇓ | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||
| H2O
PPi |
Nukleosidtriphosphat- Diphosphatase | 3.6.1.19 | Hyd | ||||||||
|
|||||||||||


Von der RNA über Nukleotid und Nukleosid zur Base und vice versa
Bearbeiten| All. | ⇓ | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||
| PPi
RNAn |
PPi
RNAn |
DNA-abh. RNA-Polymerase | 2.7.7.6 | Tr | |||||||
|
|||||||||||
| H2O
Pi |
1. |
AMP
ADP |
Ca | 1) Apyrase | 3.6.1.5 | Hyd | |||||
| 2) Nukleosidtriphosphat--Adenylat-Kinase | 2.7.4.10 | Tr | |||||||||
|
ATP |
oder
|
ATP |
oder
Nukleosid-diphosphat-Kinase |
2.7.4.6 | Tr | ||||||
|
|||||||||||
| H2O
Pi |
/ | Nukleosid-diphosphatase | 3.6.1.6 | Hyd | |||||||
| Ca | Apyrase | 3.6.1.5 | |||||||||
|
ATP |
oder
|
ATP |
oder
Cytidylat-Kinase |
2.7.4.14 | Tr | ||||||
|
|||||||||||
| H2O
Pi |
1. |
ADP
ATP |
1) 5'-Nukleotidase | 3.1.3.5 | Hyd | ||||||
| 2) Uridin-Kinase | 2.7.1.48 | Tr | |||||||||
|
|||||||||||
| Pi | Pi | Uridin-Phosphorylase | 2.4.2.3 | Tr | |||||||
|
|||||||||||

Endgültiger Abbau von Uracil
Bearbeiten| All. | ⇓ | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||
| NADPH/H+ | NADPH/H+ | Dihydropyrimidin-Dehydrogenase (NADP+) | 1.3.1.2 | Ox | DPD-Defizienz | ||||||
|
|||||||||||
| H2O
|
H2O
|
Dihydropyrimidinase | 3.5.2.2 | Hyd | DPYS-Defizienz | ||||||
|
|||||||||||
| H2O
CO2, NH3 |
β-Ureidopropionase | 3.5.1.6 | Hyd | UPB1-Defizienz | |||||||
|
|||||||||||


Uridin wird in 3 Schritten zu β-Alanin abgebaut: durch Reduktion, hydrolytische Ringspaltung und Abspaltung des Carbamoyl-Restes in Form von Ammoniak und Kohlenstoffdioxid. Reaktionen und beteiligte Enzyme sind identisch zum Thymin-Abbau.
Biosynthese von CTP aus UTP
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||
| + GTP
- CTP |
ATP, Glutamin
ADP, Pi, Glutamat |
CTP-Synthetase | 6.3.4.2 | Lig | |||||||
|
|||||||||||

DIE CTP-Synthetase katalysiert die ATP-abhängige Amidierung des Pyrimidin-Ringsystems am C4-Atom. Den dafür benötigten Stickstoff liefert Glutamin.
Stoffwechsel der Cytidinphosphate
BearbeitenFreisetzung von CDP aus der RNA
Bearbeiten| All. | ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||
| Pi
RNAn |
Polyribonukleotid-Nukleotidyltransferase | 2.7.7.8 | Tr | ||||||||
|
|||||||||||


Von der RNA über Nukleotid zum Nukleosid (und vice versa) und bis zur Base
Bearbeiten| All. | ⇓ | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||
| PPi
RNAn |
PPi
RNAn |
DNA-abh. RNA-Polymerase | 2.7.7.6 | Tr | |||||||
|
|||||||||||
| H2O
Pi |
Ca | Apyrase | 3.6.1.5 | Hyd | |||||||
|
ATP |
oder
|
ATP |
oder
Nukleosid-diphosphat-Kinase |
2.7.4.6 | Tr | ||||||
|
|||||||||||
| H2O
Pi |
Ca | Apyrase | 3.6.1.5 | Hyd | |||||||
|
ATP |
oder
|
ATP |
oder
Cytidylat-Kinase |
2.7.4.14 | Tr | ||||||
|
|||||||||||
| H2O
Pi |
1. |
ADP
ATP |
1) 5'-Nukleotidase | 3.1.3.5 | Hyd | ||||||
| 2) Uridin-Kinase | 2.7.1.48 | Tr | |||||||||
|
|||||||||||
| H2O
NH3 |
Cytidin-Deaminase | 3.5.4.5 | Hyd | ||||||||
|
|||||||||||
| Pi | Pi | Uridin-Phosphorylase | 2.4.2.3 | Tr | |||||||
|
|||||||||||


Uracil kann wiederverwertet oder zu β-Alanin abgebaut werden, wie oben dargestellt.
Biosynthese von dCDP aus CDP
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| Red. Thioredoxin
H2O, Thioredoxin-Disulfid |
ATP, Fe | Ribonukleosid-diphosphat- Reduktase | 1.17.4.1 | Ox | ||||||
|
||||||||||

Stoffwechsel der Desoxycytidinphosphate
BearbeitenVon der DNA über Nukleotid zum Nukleosid (und vice versa) und bis zur Base
Bearbeiten| All. | ⇓ | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||
| PPi
DNAn |
PPi
RNAn |
DNA-abh. DNA-Polymerase | 2.7.7.7 | Tr | |||||||
|
|||||||||||
| ADP
ATP |
ADP
ATP |
Nukleosid-diphosphat-Kinase | 2.7.4.6 | Tr | |||||||
|
|||||||||||
| ADP
ATP |
Cytidylat-Kinase | 2.7.4.14 | Tr | ||||||||
|
|||||||||||
| H2O
Pi |
1. |
NDP
NTP |
1) 5'-Nukleotidase | 3.1.3.5 | Hyd | ||||||
| 2) Desoxycytidin-Kinase | 2.7.1.74 | Tr | |||||||||
|
|||||||||||
| H2O
NH3 |
Cytidin-Deaminase | 3.5.4.5 | Hyd | ||||||||
|
|||||||||||
| Pi
Desoxy-α-D-Rib-1-P |
Purinnukleosid-Phosphorylase | 2.4.2.1 | Tr | PNP-Def. | |||||||
| Thymidin-Phosphorylase | 2.4.2.4 | Tr | MTDPS1 (MNGIE) | ||||||||
|
|||||||||||





Uracil kann wiederverwertet oder zu β-Alanin abgebaut werden, wie oben dargestellt.
Biosynthese von dUMP aus dCMP
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| H2O
NH3 |
dCMP-Deaminase | 3.5.4.12 | Hyd | |||||||
|
||||||||||

Biosynthese von dUDP aus UDP
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| Red. Thioredoxin
H2O, Thioredoxin-Disulfid |
ATP, Fe | Ribonukleosid-diphosphat- Reduktase | 1.17.4.1 | Ox | ||||||
|
||||||||||

Stoffwechsel der Desoxyuridinphosphate
BearbeitenVon der DNA über Nukleotid zum Nukleosid (und vice versa) und bis zur Base
Bearbeiten| All. | ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||
| ADP
ATP |
ADP
ATP |
Nukleosid-diphosphat-Kinase | 2.7.4.6 | Tr | |||||||
|
|||||||||||
| ADP
ATP |
ADP
ATP |
dTMP-Kinase (Thymidylatkinase) | 2.7.4.9 | Tr | |||||||
|
|||||||||||
| ADP
ATP |
Thymidin-Kinase | 2.7.1.21 | Tr | ||||||||
|
|||||||||||
| Pi
Desoxy-α-D-Rib-1-P |
Purinnukleosid-Phosphorylase | 2.4.2.1 | Tr | PNP-Def. | |||||||
| Thymidin-Phosphorylase | 2.4.2.4 | Tr | MTDPS1 (MNGIE) | ||||||||
|
|||||||||||

Uracil kann wiederverwertet oder zu β-Alanin abgebaut werden, wie oben dargestellt.
Biosynthese von dTMP aus dUMP
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| N5,N10-Methylen-THF |
|
2.1.1.45 | Tr | |||||||
|
||||||||||

Stoffwechsel der Desoxythymidinphosphate
BearbeitenVon der DNA über Nukleotid zum Nukleosid (und vice versa) und bis zur Base
Bearbeiten| All. | ⇓ | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||
| PPi
DNAn |
PPi
RNAn |
DNA-abh. DNA-Polymerase | 2.7.7.7 | Tr | |||||||
|
|||||||||||
| H2O
Pi |
Ca | Apyrase | 3.6.1.5 | Hyd | |||||||
|
ATP |
oder
|
ATP |
oder
Nukleosid-diphosphat-Kinase |
2.7.4.6 | Tr | ||||||
|
|||||||||||
| H2O
Pi |
Ca | Apyrase | 3.6.1.5 | Hyd | |||||||
|
ATP |
oder
|
ATP |
oder
dTMP-Kinase (Thymidylat-Kinase) |
2.7.4.9 | Tr | ||||||
|
|||||||||||
| H2O
Pi |
1. |
ADP
ATP |
1) 5'-Nukleotidase
2) Thymidinkinase |
3.1.3.5 | Hyd
Tr |
||||||
|
|||||||||||
| Pi
Desoxy-α-D-Rib-1-P |
Thymidin-Phosphorylase | 2.4.2.4 | Tr | MTDPS1 (MNGIE) | |||||||
|
|||||||||||




Endgültiger Abbau von Thymin
Bearbeiten| All. | ⇓ | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||
| NADPH/H+ | NADPH/H+ | Dihydropyrimidin-Dehydrogenase (NADP+) | 1.3.1.2 | Ox | DPD-Defizienz | ||||||
|
|||||||||||
| H2O
|
H2O
|
Dihydropyrimidinase | 3.5.2.2 | Hyd | DPYS-Defizienz | ||||||
|
|||||||||||
| H2O
CO2, NH3 |
β-Ureidopropionase | 3.5.1.6 | Hyd | UPB1-Defizienz | |||||||
|
|||||||||||


Der Abbau von Thymin erfolgt analog zum Uracil-Abbau (vgl. oben).
Biologische Bedeutung der Pyrimidinnukleotide
BearbeitenFür die RNA-Biosynthese werden die Pyrimidinnukleotide CTP und UTP benötigt (Uracil ersetzt in der RNA das Thymidin der DNA), für die Synthese von DNA braucht die Zelle die Desoxynukleotide dCTP und dTTP. UTP wird zusätzlich für die Aktivierung von Glucose zu UDP-Glucose benötigt, z.B. für die Biosynthese von Glycogen und Glucuronsäure sowie den Galactose-Stoffwechsel. UTP aktiviert auch den Aminozucker N-Acetyl-D-Glucosamin-1-P zu UDP-N-Acetyl-D-Glucosamin. Analog wird Cholin mit CTP zu CDP-Cholin aktiviert, z.B. für die Biosynthese von Lecithin und der Aminozucker N-Acetylneuraminat (NANA) zu CMP-N-Acetylneuraminat.
Pharmakologie
Bearbeiten
Dihydrofolat-Reduktase-Hemmer wie Methotrexat (MTX) hemmen den Folat-Stoffwechsel global, indem sie die Bildung der biologisch aktiven Tetrahydrofolsäure (THF) aus Folsäure und Dihydrofolsäure (DHF) unterbinden (siehe dort). Dadurch kommt es zur Hemmung sowohl der Purin-Synthese als auch der Pyrimidin-Synthese (Thymidylat-Synthase). Die Depletion an Nukleotiden führt in der Folge zur Reduktion der Zellvermehrung, so dass über diesen Mechanismus z.B. die Proliferation von Immun- oder Krebszellen gebremst werden kann.
Eine selektive Blockade der Pyrimidin-Synthese ist mit Leflunomid möglich. Das Immunsuppressivum hemmt die Dihydroorotat-Dehydrogenase, die an der UMP-Biosynthese beteiligt ist. Als Basistherapeutikum wird es in der Behandlung von rheumatischen Erkrankungen angewandt.
Die Pyrimidin-Synthese kann weiterhin mit verschiedenen Pyrimidin-Analoga blockiert werden. Ein Beispiel ist 5-Fluoruracil (5-FU). Aufgrund seiner Ähnlichkeit mit den Nukleinbasen Uracil, Cytosin und Thymin inhibiert 5-FU die Thymidylat-Synthase. Eingesetzt wird es als Chemotherapeutikum v.a. bei Darmkrebs und Brustkrebs, sowie äußerlich bei Warzen.
Literatur
BearbeitenWeblinks
Bearbeiten
Aminosäuren-Stoffwechsel
Bearbeiten
Allgemeines
BearbeitenSämtliche Proteine sind aus 20 (21+) α-Aminosäuren aufgebaut. Dabei werden nur die L-Formen der α-Aminosäuren verwendet. Etwa die Hälfte dieser Aminosäuren ist für die meisten Tiere, und somit auch den Menschen, essentiell und muss mit der Nahrung aufgenommen werden. Neben der Proteinbiosynthese werden die Aminosäuren für die Bildung zahlreicher Funktionsmoleküle und Signalstoffe verwendet.
Die Biosynthese der essentiellen proteinogenen L-Aminosäuren
Bearbeiten| Aminosäure | Ursprung | Enzyme | Quelle |
|---|---|---|---|
| Threonin | Aspartat | 5 | KEGG, WP |
| Lysin | Aspartat | 6 - 9 | KEGG |
| Methionin | Aspartat und Cystein | 7 (+ 2 für Cystein) | KEGG, KEGG, WP |
| Phenylalanin | Erythrose-4-phosphat (HMP-Weg) und PEP (Glycolyse) | 10 | KEGG |
| Tryptophan | Erythrose-4-phosphat (HMP-Weg) und PEP (Glycolyse) | 12 | KEGG |
| Histidin | PRPP (HMP-Weg) | 10 | KEGG |
| Valin | Pyruvat | 6 | KEGG |
| Leucin | Pyruvat | 5 | KEGG |
| Isoleucin | Pyruvat (und Threonin) | 10 (- 12) | KEGG |
Zu den essentiellen Aminosäuren gehören Threonin, Lysin und Methionin, die aromatischen Aminosäuren Phenylalanin, Tryptophan und Histidin sowie die verzweigtkettigen Aminosäuren Valin, Leucin und Isoleucin. Der Körper hat aufgrund des ausreichenden Angebots in der Nahrung die Enzyme für die Biosynthese dieser Aminosäuren verloren. Die Frage, warum sich gerade bei diesen Aminosäuren das „Outsourcing“ gelohnt haben könnte, lässt sich beantworten, wenn man sich die Synthesewege anschaut. Diese sind gerade bei den essentiellen Aminosäuren besonders aufwendig (viele Reaktionsschritte) und energieintensiv.
Pflanzen produzieren sich wegen den limitierten Möglichkeiten der Nahrungsaufnahme und der einfachen Energieversorgung (Photosynthese) auch weiterhin aus Glucose alle Aminosäuren und alle anderen zum Leben notwendigen komplexeren Moleküle selbst.
Die Biosynthese der nicht-essentiellen proteinogenen Aminosäuren
Bearbeiten
| ||||||||||||||||||||||||||||
| Farbcode: Stickstoffaufnahme/-abgabe | ||||||||||||||||||||||||||||
Die Synthesewege der nicht-essentiellen Aminosäuren sind sehr übersichtlich. Sie beinhalten nur wenige Reaktionsschritte und zweigen von der Glycolyse, dem Citratzyklus und dem Harnstoffzyklus ab.
Zuerst werden die α-Aminosäuren Serin, Alanin, Aspartat und Glutamat durch Aminierung oder Transaminierung (Stickstoffaufnahme bzw. -übertragung) am Cα-Atom der entsprechenden α-Ketosäuren gebildet, die aus Glycolyse und Citratzyklus stammen. Davon leiten sich weitere Aminosäuren ab: Glycin aus Serin, Asparagin aus Aspartat sowie Glutamin, Prolin und Arginin aus Glutamat. Damit wäre bereits die Biosynthese von 9 proteinogenen Aminosäuren abgedeckt. 3 weitere - Tyrosin, Cystein und Selenocystein - gehen aus den essentiellen Aminosäuren Phenylalanin und Methionin hervor.
Im Einzelnen erfolgt die Synthese wie folgt:
- Glycin und Serin werden aus 3-Phosphoglycerat (Glycolyse) synthetisiert. 3-Phosphoglycerat wird zuerst zur α-Ketosäure 3-Phosphohydroxypyruvat oxidiert und dann Pyridoxalphosphat-abhängig zu Serin transaminiert. Aus Serin wird Glycin gebildet.
- Alanin wird durch Pyridoxalphosphat-abhängige Transaminierung aus Pyruvat gebildet.
- Aspartat entsteht durch Pyridoxalphosphat-abhängige Transaminierung aus Oxalacetat (Citratzyklus). Aus Aspartat kann Asparagin hergestellt werden.
- Glutamat wird durch Aminierung oder Pyridoxalphosphat-abhängige Transaminierung aus α-Ketoglutarat (Citratzyklus) gebildet. Aus Glutamat können dann Glutamin, Prolin und über den Harnstoffzyklus Arginin synthetisiert werden.
- Das schwefelhaltige Cystein entsteht aus Serin beim Abbau der schwefelhaltigen essentiellen Aminosäure Methionin (siehe hier). Aus Serin kann Selenocystein gebildet werden, eine seltene Aminosäure, die im genetischen Code kein eigenes Codon besitzt und deswegen gegenüber den 20 herkömmlichen proteinogenen Aminosäuren einen gewissen Sonderstatus hat.
- Tyrosin wird aus der essentiellen Aminosäure Phenylalanin generiert (siehe hier).
Abbau der proteinogenen L-Aminosäuren
Bearbeiten| Aminosäure | Ketogene Produkte | Glucogene Produkte |
|---|---|---|
| Alanin | Pyruvat | |
| Glycin -> Serin | Pyruvat | |
| Threonin | Acetaldehyd | Pyruvat, Succinyl-CoA |
| Cystein | Pyruvat | |
| Asparagin -> Aspartat | Oxalacetat / Fumarat | |
| Glutamin -> Glutamat | α-Ketoglutarat | |
| Prolin -> Glutamat | α-Ketoglutarat | |
| Arginin -> Glutamat | α-Ketoglutarat | |
| Histidin -> Glutamat | α-Ketoglutarat | |
| Methionin | Succinyl-CoA | |
| Lysin | Acetyl-CoA | |
| Phenylalanin -> Tyrosin | Acetoacetat | Fumarat |
| Tryptophan | Acetyl-CoA | Pyruvat |
| Valin | Succinyl-CoA | |
| Leucin | Acetoacetat, Acetyl-CoA | |
| Isoleucin | Acetyl-CoA | Succinyl-CoA |
| Farbcode: Essentielle Aminosäuren | ||
Glucogene Aminosäuren werden zu Pyruvat oder zu den Intermediaten des Citratzyklus (Oxalacetat, α-Ketoglutarat, Succinyl-CoA, Fumarat) abgebaut und können zur Gluconeogenese herangezogen werden. Ketogene Aminosäuren werden zu Acetaldehyd, zum Ketonkörper Acetoacetat oder direkt zu Acetyl-CoA degradiert.
In den meisten Fällen steht am Anfang des Aminosäurenabbaus die Abspaltung der Aminogruppe durch Transaminierung. Der Stickstoff wird über Glutamat und Aspartat in den Harnstoffzyklus eingeschleust und als Harnstoff oder direkt als Ammoniak über die Niere ausgeschieden.
Posttranslationale Modifikation der proteinogenen Aminosäuren
BearbeitenNachdem die L-Aminosäuren in Proteine eingebaut wurden können sie durch kovalente Modifikation weiter verändert werden. Damit lassen sich die Eigenschaften und Funktionen eines Proteins auch noch nach seiner Fertigstellung verändern. Bespiele:
- Die Hydroxyl-Gruppen der Serin-, Threonin- und Tyrosin-Reste können phosphoryliert oder mit Glycanen (O-Glycosylierung) versehen werden. Die Phosphorylierung und die dadurch bewirkte Konformationsänderung des Proteins wird z.B. für das An- und Abschalten von Enzymen (sog. Interkonversion) verwendet.
- Die γ-Amid-Gruppe von Asparagin kann ebenfalls glycosyliert werden (N-Glycosylierung).
- Histidin kann zum 3-Methylhistidin metyliert werden.
- Prolin und Lysin können hydroxyliert werden, so dass γ-Hydroxyprolin und δ-Hydroxylysin entsteht. Das findet man z.B. bei der Kollagen-Synthese.
- Glutamat kann in der γ-Position carboxyliert werden. Die γ-Carboxylierung ist z.B. bei der posttranslationalen Modifikation der Vitamin K-abhängigen Gerinnungsfaktoren anzutreffen.
Nicht-proteinogene Aminosäuren
BearbeitenIm Intermediarärstoffwechsel fallen auch zahlreiche nicht-proteinogene Aminosäuren an. Beispiele sind die α-Aminosäuren Ornithin (Harnstoffzyklus), Homocystein (Methionin-Abbau), 5-Hydroxy-Tryptophan (Serotonin-Biosynthese), die β-Aminosäure β-Alanin (Uracil-Abbau) und die γ-Aminosäure γ-Aminobuttersäure (GABA, Glutamat-Stoffwechsel).
Rolle der Aminosäuren für die Zelle bzw. den Organismus
Bearbeiten
- Die proteinogenen L-Aminosäuren sind die Bausteine sämtlicher Proteine (Enzyme, Strukturproteine usw.). Dafür werden die Aminosäuren am Ribosom zu einer langen Peptid-Kette verknüpft. Die Peptidbindung zwischen zwei Aminosäuren entsteht, indem die Amino-Gruppe der einen Aminosäure mit der Carboxyl-Gruppe der nächsten Aminosäure unter Wasserabspaltung reagiert.
- Die Abbauprodukte der Aminosäuren - aus der Nahrung oder aus dem körpereigenen Proteinbestand - können zur Energiegewinnung und zur Synthese von Fettsäuren und Cholesterin genutzt werden und die glucogenen Aminosäuren zusätzlich auch zum Wiederauffüllen des Citratzyklus zur Gluconeogenese.
- Im Vielzeller fungieren viele Aminosäuren (Glutamat, Glycin, Aspartat) und ihre Derivate (GABA, L-Thyroxin, Dopamin, Adrenalin und Noradrenalin, Serotonin und Histamin) als Transmittersubstanzen bzw. Hormone.
- Aus oder unter Beteiligung von Aminosäuren werden weitere wichtige Moleküle gebildet wie z.B. Carnitin (aus Lysin), Taurin (aus Cystein), Kreatin (aus Arginin und Glycin), Häm (mit Glycin), Coenzym A (mit Cystein) oder Sphingolipide (mit Serin).
Glutamat - Die Drehscheibe des Stickstoff-Stoffwechsels
BearbeitenStickstoff in Form von Ammoniak (NH3) oder Aminogruppen (-NH2) kann im Stoffwechsel reversibel auf Pyruvat, Oxalacetat und α-Ketoglutarat übertragen werden, die aus der Glycolyse und dem Citratzyklus stammen. Dadurch werden Alanin, Glutamat (und Glutamin) und Aspartat gebildet. Dies hat mehrerlei Bedeutung:
- Beseitigung von freiem (giftigen) Ammoniak, das insbesondere aus dem Aminosäurenabbau stammt.
- Verwendung des fixierten Stickstoffs für die Biosynthese von Aminosäuren, Harnstoff (zur Stickstoffausscheidung) und anderen stickstoffhaltigen Verbindungen wie Purine und Pyrimidine.
- Durch die Umkehrung der Aminogruppenübertragung können die genannten Aminosäuren (durch die Nahrung aufgenommen oder durch Muskelabbau in Hungerzeiten freigesetzt) entsprechend Substrat liefern zur Energieerzeugung oder zur Gluconeogenese, sowie zur Wiederauffüllung des Citratzyklus mit C4-Körpern.
Der Stickstoff aus freiem Ammoniak wird zuerst im Glutamat fixiert und dann auf weitere Moleküle übertragen. Glutamat nimmt daher eine zentrale Stellung ein. Als Coenzym der Transaminasen dient häufig Pyridoxalphosphat (PLP, Vitamin B6). Das Vitamin ist auch an vielen anderen Reaktionen im Aminosäurenstoffwechsel beteiligt.
| Reaktionen | Enzym | Abk. | ||
|---|---|---|---|---|
| α-Ketoglutarat + NH3 | ⇔ | Glutamat | Glutamatdehydrogenase | GLDH |
| Glutamat + NH3 | ⇔ | Glutamin | Glutaminsynthetase | |
| Glutamat + Pyruvat | ⇔ | α-Ketoglutarat + Alanin | Glutamat-Pyruvat-Transaminase bzw. Alanin-Aminotransferase | GPT bzw. ALAT |
| Glutamat + Oxalacetat | ⇔ | α-Ketoglutarat + Aspartat | Glutamat-Oxalacetat-Transaminase bzw. Aspartat-Aminotransferase | GOT bzw. ASAT |
Über die Verstoffwechselung von Aminosäuren gelangt Stickstoff auch in andere Stoffwechselwege. Dabei dienen insbesondere Glutamat, Glutamin und Aspartat als Stickstoff-Donatoren. Der Amino-Stickstoff wird benötigt u.a. für:
- Aminozucker-Stoffwechsel (Glutamin)
- Porphyrin-Biosynthese (Glycin)
- Purin-Biosynthese (Glycin, Glutamin, Aspartat)
- Pyrimidin-Stoffwechsel (Glutamin, Aspartat)
- Glycin- und Serinbiosynthese (Glutamat)
- Harnstoffbiosynthese (Glutamat, Aspartat)
- Kreatin-Biosynthese (Glycin, Arginin)
- Sphingolipid-Biosynthese (Serin)
Ausgeschieden wird der Stickstoff in Form von Harnstoff, der v.a. in der Leber gebildet wird und den Körper über die Niere verlässt.
Klinische Chemie und Laboratoriumsmedizin
BearbeitenDa die Enzyme GOT, GPT und GLDH überwiegend in der Leber vorkommen kann ihre Bestimmung im Blutserum zur Diagnostik von Leberschädigungen verwendet werden. Die GPT kommt fast ausschließlich im Zytoplasma vor, die GOT zu 30 % im Zytoplasma und zu 70 % in den Mitochondrien, die GLDH ausschließlich in den Mitochondrien. Daher kann aus dem Muster der Werte auf das Ausmaß der Zellschädigung rückgeschlossen werden. Dabei bestimmt man gerne den Ritis-Quotient = GOT/GPT. Bei leichten Zellschäden ist v.a. die GPT erhöht (Ritis-Q. < 1), bei schweren Schäden mit Beeinträchtigung der Zellorganellen dominieren die GOT und GLDH (Ritis-Q. > 1). Diese Parameter bieten aufgrund ihrer kurzen Halbwertszeit (GOT 17 h, GPT 47 h) und der guten Korrelation zwischen Schadensausmaß und Spiegelhöhe eine gute Momentaufnahme und sind gut zur Verlaufsbeobachtung geeignet. Die GOT kann auch bei Herzinfarkt oder muskulären Schäden erhöht sein.
Weblinks: Laborlexikon: GPT, GOT, GLDH
Weblinks
Bearbeiten
Glutamat- und Glutamin-Stoffwechsel
BearbeitenAllgemeines
BearbeitenL-Glutamat (L-Glutaminsäure) ist eine nicht-essentielle, proteinogene Aminosäure mit einer sauren, hydrophilen Carboxylgruppe-tragenden Seitenkette. Die α-Aminosäure Glutamat bzw. die zugehörige α-Ketosäure α-Ketoglutarat hat im Stoffwechsel eine herausragende Rolle als Stickstoff-Sammel- und Verteilungsstelle. Glutamat und das davon abgeleitete biogene Amin γ-Aminobuttersäure (GABA) wirken weiterhin im ZNS als Neurotransmitter.
L-Glutamin ist eine nicht-essentielle, proteinogene Aminosäure mit einer hydrophilen Säureamidgruppe-tragenden Seitenkette. Sie dient ebenfalls als Vehikel für Aminogruppen.
Glutamat und Glutamin
Bearbeiten| ⇓ | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| NH3, NAD(P)H/H+
H2O, NAD(P)+ |
NAD(P)H/H+, NH3
NAD(P)+, H2O |
Glutamat-Dehydrogenase (GLDH) | 1.4.1.3 | Ox | GLUD1-Def. (Hyperinsulinismus- Hyperammonämie-S. (HHS)) | |||||
|
||||||||||
| NH3, ATP
ADP, Pi |
1. |
NH3 H2O |
|
6.3.1.2 | Lig | GLNS-Def. | ||||
| 2) Glutaminase | 3.5.1.2 | Hyd | ||||||||
|
||||||||||


α-Ketoglutarat reagiert mit Ammoniak oder einer Aminogruppe zu Glutamat. Dies ist die wichtigste reversible Reaktion, über die der Stickstoff fixiert wird, der in verschiedenen Stoffwechselreaktionen, v.a. aber beim Abbau von Aminosäuren und deren Derivaten, frei wird. Durch Aufnahme einer weiteren Stickstoffgruppe entsteht aus Glutamat - unter ATP-Verbrauch - Glutamin. Glutamat verteilt den Stickstoff nun weiter, z.B. auf Pyruvat (Alanin-Biosynthese) und Oxalacetat (Aspartat-Biosynthese). Die entsprechenden Enzymaktivitäten (GOT, GPT, GLDH) sind besonders in der Leber ausgeprägt, da hier auch größtenteils die Harnstoffsynthese stattfindet.
Glutamat, Glutamin und Aspartat schleusen den Stickstoff nun in weitere Stoffwechselwege ein, entweder durch Übertragung der Aminogruppe oder komplexere chemische Reaktionen. Dazu gehören z.B.:
- Alanin-Bildung (Glu)
- Biosynthese von Aspartat (Glu) und Asparagin aus Aspartat (Gln).
- Harnstoffzyklus in der Leber (Glu, Gln, Asp) mit Bildung von Harnstoff oder Arginin. Letzteres ist Ausgangspunkt der Biosynthese von Creatin und basischer Polyamine.
- Aus Glutamat werden Ornithin (L-AS) und Prolin gebildet.
- Biosynthese von Serin und Glycin (Glu).
- Glutathion-Biosynthese (Glu).
- Proteinbiosynthese (Translation) (Glu, Gln, Asp, Ala u.a.m.).
- Aminozucker-Stoffwechsel (Gln)
- Purin-Biosynthese (Gln, Asp).
- Pyrimidin-Stoffwechsel (Gln, Asp).
Glutamin ist unter den Aminosäuren diejenige mit der höchste Plasmakonzentration. Neben Alanin ist Glutamin für den Stickstoff-Transport über den Blutweg verantwortlich. Glutamin kann so z.B. Ammoniak vom Gehirn über den Blutweg Richtung Niere transportieren. In der Niere kann Glutamin durch die mitochondriale Glutaminase-Reaktion Ammoniak freisetzen, das zusammen mit einem Proton (d.h. Säure) ausgeschieden wird. Dadurch wird im Harnstoffzyklus Bicarbonat eingespart (pH-Regulation).
Der Abbau von Glutamin und Glutamat liefert wieder α-Ketoglutarat, das nach Umwandlung in Oxalacetat (Citratzyklus) für die Gluconeogenese genutzt werden kann. Glutamin ist das bevorzugte Substrat für die Gluconeogenese in der Niere.
Weitere Informationen zu den einzelnen Aminosäuren findet man auf deren Unterseiten.
Glutamat spielt eine wichtige Rolle als exzitatorischer Neurotransmitter im ZNS (und in der Nahrungsmittelindustrie als Geschmacksverstärker, Geschmacksrichtung Umami). Die Decarboxylierung von Glutamat führt zum biogenen Amin γ-Aminobuttersäure (GABA), ein ebenfalls wichtiger aber inhibitorischer Neurotransmitter im Gehirn:
γ-Aminobuttersäure
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
|
|
Pyridoxal- phosphat | Glutamat-Decarboxylase | 4.1.1.15 | Ly | GAD1-Def. (spastic cerebral palsy) | |||||
|
||||||||||
| α-Ketoglutarat | α-Ketoglutarat | GABA-Transaminase (ABAT) | 2.6.1.19 | Tr | ABAT-Def. | |||||
|
||||||||||
| H2O, NAD+ | Succinat-Semialdehyd- Dehydrogenase | 1.2.1.24 | Ox | SSADH-Def. | ||||||
|
||||||||||
GABA wird zu Succinat abgebaut, das mit CoA-SH aktiviert und wieder in den Citratzyklus eingeschleust werden kann.
Weblinks
Bearbeiten
Alanin-Stoffwechsel
BearbeitenAllgemeines
BearbeitenL-Alanin ist eine nicht-essentielle, kleine, proteinogene Aminosäure mit einer aliphatischen, unpolaren, hydrophoben Seitenkette.
Biosynthese und Abbau von Alanin durch Transaminierung
Bearbeiten| ⇓ | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
| Glutamat | Glutamat | Pyridoxal- phosphat | Alanin-Transaminase (ALT, ALAT, GPT) | 2.6.1.2 | Tr | ||


Biologische Bedeutung
Bearbeiten- Biosynthese von Alanin z.B. für die Proteinbiosynthese.
- Abbau von Alanin zu Pyruvat z.B. für die Gluconeogenese oder den Citratzyklus.
- Glucose-Alanin-Zyklus (siehe unter Glycolyse).
- Alanin entsteht (neben Acetyl-CoA) auch beim Abbau der essentiellen Aminosäure Tryptophan.
Weblinks
Bearbeiten
Aspartat- und Asparagin-Stoffwechsel (Aspartatzyklus)
BearbeitenAllgemeines
BearbeitenL-Aspartat (L-Asparaginsäure) ist eine nicht-essentielle, proteinogene Aminosäure mit einer hydrophilen, sauren Carboxylgruppe in der Seitenkette. Die Aminosäure entsteht aus Oxalacetat durch Übernahme einer Stickstoff-Gruppe von Glutamat. Aspartat wird u.a. benötigt für die Purin-, Pyrimidin- und die Harnstoffsynthese.
L-Asparagin ist eine nicht-essentielle, proteinogene Aminosäure mit einer hydrophilen Säureamid-tragenden Seitenkette. Sie leitet sich vom Aspartat ab.
Biosynthese und Abbau von Aspartat und der Aspartatzyklus
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| L-Glutamat | L-Glutamat | Pyridoxal- phosphat | Aspartat-Transaminase (AST, ASAT, GOT) | 2.6.1.1 | Tr | |||||
|
||||||||||
| NTP, R=O
NDP + Pi od. NMP + PPi |
Y-Synthetase | Lig | ||||||||
|
||||||||||
|
|
Y-Lyase | Ly | ||||||||
|
||||||||||
| H2O
|
H2O
|
Fumarase 1 | 4.2.1.2 | Ly | Fumarase-Def., HLRCC, MCUL1 | |||||
|
||||||||||
| NAD+ | NAD+ | Malat-Dehydrogenase | 1.1.1.37 | Ox | ||||||
|
||||||||||

Die Aspartat-Transaminase kann sowohl zur Erzeugung von Aspartat genutzt werden als auch zu dessen Abbau, z.B. um Oxalacetat für die Gluconeogenese freizusetzen. Das Enzym wirkt außerdem am Malat-Aspartat-Shuttle mit, ein System zum Transport von Reduktionsäquivalenten in das Mitochondrium.
Der Aspartatzyklus läuft im Zytosol ab einschließlich der Teilschritte des Citratzyklus. Er liefert den aus dem Glutamat stammenden Stickstoff in folgende Wege:
Als ganzes geht Aspartat ein in die:
- Pyrimidin-Biosynthese
- Proteinbiosynthese
Eine physiologische Rolle hat Aspartat im ZNS als Neurotransmitter.
Biosynthese von Asparagin
BearbeitenAspartat (Asparaginsäure) kann von Glutamin unter ATP-Hydrolyse eine weitere Stickstoffgruppe übernehmen und wird zu Asparagin:
| All. | ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||
| - Glutamin | H2O, ATP, L-Glutamin
AMP, PPi, L-Glutamat |
L-Glutamin, ATP, H2O
L-Glutamat, AMP, PPi |
Asparagin-Synthase | 6.3.5.4 | Lig | ||||||
|
|||||||||||
In Glykoproteinen bildet Asparagin eine Zuckeranbindungsstelle (N-Glykosylierung; Anm.: N steht hier nicht für Stickstoff, sondern für Asparagin).
Weblinks
Bearbeiten
Harnstoffzyklus
BearbeitenAllgemeines
BearbeitenDer Harnstoffzyklus findet überwiegend in der Leber statt und zwar verteilt auf Zytosol und Mitochondrium. Er dient der Entgiftung von Ammoniak, das insbesondere beim Proteinabbau frei wird. 1932 wurde er von Hans Adolf Krebs und Kurt Henseleit entdeckt. Es war damit der erste metabolische Zyklus, der beschrieben wurde.
Neben Harnstoff wird im Harnstoffzyklus auch die Aminosäure Arginin gebildet.
Bildung von Carbamoylphosphat im Mitochondrium
Bearbeiten| All. | ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||
| + N-Acetyl- glutamat | 2 ATP, NH3
2 ADP, Pi |
Carbamoylphosphat-Synthetase I Mitochondrium | 6.3.4.16 | Lig | Hyperammonämie I | ||||||
|
|||||||||||
Die Carbamoyl-phosphat-Synthetase I wird allosterisch aktiviert von N-Acetylglutamat. Dieses dient dem Enzym als Parameter bzw. Sensor für das Glutamat- und damit für das Ammoniak-Vorkommen.
Der Harnstoffzyklus
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| Carbamoylphosphat
Pi |
Ornithin-Carbamoyltransferase Mitochondrium | 2.1.3.3 | Tr | Hyperammonämie II | ||||||
|
||||||||||
| Citrullin-Transporter Innere Mitochondrienmembran |
Citrullin-Transporter-Defizienz | |||||||||
| ATP, Aspartat
AMP + PPi |
Argininosuccinat-Synthase Zytosol |
6.3.4.5 | Lig | Citrullinämie I | ||||||
|
||||||||||
| Argininosuccinat-Lyase Zytosol |
4.3.2.1 | Ly | Argininbernsteinsäure-krankheit | |||||||
|
||||||||||
|
Mn | Arginase Zytosol |
3.5.3.1 | Hyd | Argininämie | |||||
|
||||||||||
| Mitochondrialer Ornithintransporter 1 Innere Mitochondrienmembran |
Hyperornithinämie-Hyperammonämie-Homocitrullinurie-Syndrom | |||||||||




Die Reaktionen im Detail
BearbeitenDer Harnstoffzyklus ist in der Zelle auf zwei Kompartimente verteilt. Die Biosynthese von Carbamoylphosphat und die Übertragung des Carbamoyl-Restes auf Ornithin zur Bildung von Citrullin findet im Mitochondrium statt, die restlichen Reaktionen laufen im Zytosol ab. Die für die Transportprozesse zuständigen Ornithin- und Citrullintransporter sitzen in der inneren Mitochondrienmembran. Der Stickstoff, der in das Harnstoff-Molekül eingebaut wird, wird von Carbamoylphosphat und Aspartat in den Zyklus eingebracht.
- Carbamoylphosphat wird von der Carbamoylphosphat-Synthetase I im Mitochondrium aus bzw. mit Hilfe von Hydrogencarbonat, Ammoniak und ATP gebildet. Die Biosynthese wird von N-Acetylglutamat allosterisch aktiviert.
- Ebenfalls im Mitochondrium erfolgt die Übertragung des Carbamoyl-Restes unter Phosphat-Abspaltung auf Ornithin. Dabei entsteht Citrullin, das von einem spezifischen Transporter ins Zytosol verbracht wird.
- Im Zytosol wird über den Aspartatzyklus ein weiteres Stickstoff-Atom auf Citrullin übertragen, so dass aus dem endständigen Harnstoff-Rest eine Guanidino-Gruppe wird. Im Einzelnen wird Citrullin unter ATP-Verbrauch mit Aspartat zu L-Argininosuccinat kondensiert und letzteres dann in Arginin und Fumarat gespalten.
- Im letzten Schritt wird Arginin zu Ornithin und Harnstoff hydrolysiert. Nach Rücktransport ins Mitochondrium steht Ornithin für den nächsten Zyklus bereit.
Die Synthese eines Harnstoffmoleküls kostet mindestens 4 energiereiche Phosphatbindungen, was der Hydrolyse von 4 ATP zu 4 ADP + 4 Pi entspricht. Allerdings liefert der anhängende Aspartatzyklus auch ein NADH/H+, entsprechend 2,5 ATP.
Bedeutung für den Organismus
BearbeitenBezogen auf den Organismus spielt sich die Harnstoff-Biosynthese hauptsächlich in der Leber ab. Seine Hauptfunktion ist die Entgiftung von Ammoniak unter Bicarbonatverbrauch zu ungiftigem Harnstoff, der dann über die Niere ausgeschieden werden kann, d.h. Stickstoffausscheidung. Der Stickstoff wird in Form von Ammoniak aus der „Stickstoffsammelstelle“ Glutamat freigesetzt (Glutamat-Dehydrogenase-Reaktion), das diesen seinerseits von Alanin, Aspartat oder beim Abbau anderer Aminosäuren übernommen hat.
Stickstoff kann auch direkt in den Urin ausgeschieden werden. Dabei wird in der Niere Ammoniak aus Glutamin freigesetzt (Glutaminase-Reaktion). Das basische Ammoniakmolekül (pK 9) bindet bei den üblichen pH-Werten in der Niere generell ein Proton und wandert als Ammoniumion NH4+ in den Urin.
Da die Glutaminase-Reaktion nun einerseits Protonen zur Ausscheidung bringt und andererseits Bicarbonat (Carbamoylphosphatsynthese) einspart, kann der Organismus über das Gleichgewicht dieser beiden Stickstoffausscheidungssysteme effektiv die Protonenausscheidung und damit den Säuren-Basen-Haushalt regulieren.
Wie kommt das Ammoniak, das v.a. beim Abbau von Aminosäuren (dabei u.a. im Purinnukleotidzyklus) freigesetzt wird, zur Entgiftung in die Leber? Die wichtigste Rolle übernimmt hier Alanin, das über den Glucose-Alanin-Zyklus den Stickstoff in die Leber transportiert und dort wieder auf Glutamat überträgt. (Das bei der Transaminierung frei werdende Pyruvat kann dann direkt zur Gluconeogenese verwendet werden, die meist mit der gesteigerten Proteolyse bei kataboler Stoffwechsellage einhergeht.) Von untergeordneter Bedeutung dürfte der N-Transport via Glutamin sein.
Neben der Ammoniak-Ausscheidung dienen die Reaktionenschritte des Harnstoffzyklus auch der Biosynthese der proteinogenen Aminosäure Arginin.
Pathobiochemie
BearbeitenStörungen des Harnstoffzyklus äußern sich in der Trias Hyperammonämie, Enzephalopathie und Säure-Basen-Störungen. Als Ursachen kommen die seltenen genetischen Transporter- und Enzymdefekte in Betracht und erworbene Lebererkrankungen wie Leberzirrhose oder akutes Leberversagen z.B. durch Knollenblätterpilz- oder Paracetamolvergiftung.
Labormedizin/Mikrobiologie
Bearbeiten
Viele Bakterien können Harnstoff mit Wasser und mittels Urease (EC 3.5.1.5) in Ammoniak und Kohlendioxid spalten. Mit einer derart gebildeten basischen Ammoniakwolke schützt sich z.B. der Magenkeim Helicobacter pylori vor der Magensäure. Mit dem Helicobacter-Urease-Test (HUT) lässt sich eine Infektion nachweisen. Dabei werden Magenbiopsien in ein Harnstoff-haltiges Nährmedium gegeben (siehe Abbildung). Sind Bakterien vorhanden, so produzieren diese Ammoniak und der Indikator färbt sich rot.
Ähnlich funktioniert der 13C-Atemtest. Hier nimmt der Patient Harnstoff zu sich, der mit dem stabilen Kohlenstoff-Isotop 13C markiert ist. Ist H. pylori im Magen vorhanden, so erscheint das 13C im CO2 der Atemluft und kann infrarotspektrometrisch gemessen werden.
Ornithin wird aus Glutamat gebildet
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| ATP
ADP |
Glutamat-5-Kinase | 2.7.2.11 | Tr | |||||||
|
||||||||||
| NADPH/H+
Pi, NADP+ |
NADPH/H+
NADP+, Pi |
Glutamat-5-semialdehyd- Dehydrogenase | 1.2.1.41 | Ox | ||||||
|
||||||||||
| L-Aminosäure
α-Ketosäure |
L-Aminosäure
α-Ketosäure |
Pyridoxal- phosphat | Ornithin-Aminotransferase | 2.6.1.13 | Tr | Ornithinemia with gyrate atrophy of the choroid and retina (HOGA) | ||||
|
||||||||||

Die Bildung von L-Glutamat-5-Semialdehyd liegt auch auf dem Weg der Prolinbiosynthese.
Ornithin wird zu Glutamat abgebaut
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||||
| α-Ketosäure
L-Aminosäure |
α-Ketosäure
L-Aminosäure |
Pyridoxal- phosphat | Ornithin-Aminotransferase | 2.6.1.13 | Tr | Ornithinemia with gyrate atrophy of the choroid and retina (HOGA) | |||||||
|
|||||||||||||
| H2O, NAD+ | H2O, NAD+ | 1-Pyrrolin-5-carboxylat- Dehydrogenase | 1.5.1.12 | Ox | Hyperprolinämie II | ||||||||
|
|||||||||||||

Der 2. Schritt ist der gleiche wie beim Abbau von Prolin. Auch Arginin, das durch Harnstoffabspaltung aus Ornithin entsteht, wird über diesen Weg zu Glutamat abgebaut.
Verbindungen des Harnstoffzyklus mit anderen Stoffwechselwegen
Bearbeiten- Glutamat- und Glutamin-Stoffwechsel
- Aspartatzyklus
- Ornithinsynthese aus Glutamat (s.o.)
- Prolin-Stoffwechsel (Ornithinsyntheseweg)
- Arginin-Stoffwechsel (Biosynthese von L-Arginin für die Proteinbiosynthese, die Bildung von NO, Polyaminen und Kreatinphosphat)
Weblinks
Bearbeiten- KEGG: Arginine and proline metabolism - Homo sapiens (human)
- The chemical logic behind... Aminoacid degradation and urea cycle by Prof. Doutor Pedro Silva.
- Google Video - Harnstoffzyklus, OE Kabarett Charité
Prolin-Stoffwechsel
BearbeitenAllgemeines
BearbeitenL-Prolin ist eine nicht-essentielle, proteinogene Aminosäure mit einer heterozyklischen Seitenkette. Sie wird aus Glutamat auf- und zu Glutamat abgebaut. Im Kollagen kommt sie als Hydroxyprolin vor.
Biosynthese von L-Prolin aus L-Glutamat
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||||||
| ATP
ADP |
Glutamat-5-Kinase | 2.7.2.11 | Tr | |||||||||||||
|
||||||||||||||||
| NADPH/H+
Pi, NADP+ |
NADPH/H+
NADP+, Pi |
Glutamat-5-semialdehyd- Dehydrogenase | 1.2.1.41 | Ox | ||||||||||||
|
||||||||||||||||
| NAD(P)H/H+ | NADPH/H+ | Pyrrolin-5-carboxylat-Reduktase | 1.5.1.2 | Ox | ||||||||||||
|
||||||||||||||||


L-Glutamat-5-semialdehyd ist auch eine Vorstufe des Ornithins.
Prolin und Hydroxyprolin (posttranslational Vitamin C-abhängig hydroxyliert) bilden neben Glycin die Kollagen-Helix auf.
Prolin wird zu Glutamat abgebaut
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| NAD(P)+ | NAD(P)+ | Pyrrolin-5-carboxylat-Reduktase | 1.5.1.2 | Ox | ||||||
|
||||||||||
| H2O, NAD+ | H2O, NAD+ | 1-Pyrrolin-5-carboxylat-Dehydrogenase | 1.5.1.12 | Ox | Hyperprolinämie II | |||||
|
||||||||||
Der 2. Schritt entspricht dem 2. Schritt des Ornithin-Abbaus.
Weblinks
Bearbeiten- KEGG: Urea cycle and metabolism of amino groups - Homo sapiens (human)
- KEGG: Arginine and proline metabolism - Homo sapiens (human)
Arginin-Stoffwechsel
BearbeitenAllgemeines
BearbeitenDie nicht-essentielle, basische, proteinogene Aminosäure L-Arginin stammt aus dem Harnstoffzyklus (Bildung über Ornithin aus Glutamat). Sie ist der Ausgangspunkt für die Synthese von Stickstoffmonoxid, Kreatinphosphat und basischen Polyaminen.
Biosynthese von Stickstoffmonoxid (NO) aus Arginin
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||
| m O2, n NADPH/H+
n NADP+ |
Stickoxid-Synthase | 1.14.13.39 | Ox | Infantile hypertrophische Pylorusstenose 1 (IHPS1) | |||||||
|
|||||||||||

Stickoxid (NO) - früher EDRF (endothelial derived relaxing factor) genannt - ist ein gasförmiger Mediator, der von verschiedenen Stickoxid-Synthasen (NOS) produziert wird und über die Aktivierung einer zytosolischen Guanylatcyclase (EC 4.6.1.2) besonders im venösen und koronararteriellen Gefäßbett die glatte Gefäßmuskulatur relaxiert mit dem Ergebnis einer Vasodilatation. NO bewirkt weiterhin eine Hemmung der Plättchenaggregation.
Pharmakologisch kann dieser Effekt z.B. mit Nitropräparaten imitiert und für die Behandlung von Angina pectoris, kardialem Lungenödem, arterieller und pulmonaler Hypertonie genutzt werden.
NO ist ähnlich wie reaktive Sauerstoffspezies (ROS) auch an der Immunabwehr beteiligt.
Die NO-Bildung verhält sich gegenläufig zur Glutathion-Expression/Aktivität.
Regulation der NO-Bildung
BearbeitenDie NO-Bildung wird erhöht durch:
- Lipide: Fettreiches Essen, gesättigte langkettige Fettsäuren, LDL (low-density lipoproteins), Linolensäure.
- Eisen.
Die NO-Bildung wird vermindert durch:
- Glucosamin, Taurin, n-3 PUFAs, Phytöstrogene, Polyphenole, Carotenoide, Zink.
Biosynthese von Kreatinphosphat aus Arginin und Glycin
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| L-Arginin | Glycin-Amidinotransferase | 2.1.4.1 | Tr | AGAT-Def. | ||||||
|
||||||||||
| S-Adenosylmethionin | Guanidinoacetat-N-Methyltransferase | 2.1.1.2 | Tr | GAMT-Def. | ||||||
|
||||||||||
| ATP
ADP |
ATP
ADP |
Kreatin-Kinase | 2.7.3.2 | Tr | ||||||
|
||||||||||




Kreatinphosphat bildet u.a. im Muskel ein kleines Energiedepot, das ein kurzes ATP-Defizit überbrücken kann, indem es bei Bedarf sein Phosphat auf ADP überträgt und damit ATP regeneriert.
Klinische Chemie: Das Enzym Kreatinkinase liegt in verschiedenen Isoformen vor (CK-MM, CK-MB, CK-BB). Die herzmuskelspezifische CK-MB und ihr Verhältnis zur Gesamt-CK im Blut kann zur Herzinfarktdiagnostik verwendet werden.
Das spontan entstehende Abbauprodukt des Kreatins, das Kreatinin wird in der Niere frei filtriert, nicht sezerniert und nicht rückresorbiert, daher kann der Kreatininspiegel bzw. die daraus errechnete Kreatinin-Clearance als diagnostischer Parameter für die glomeruläre Filtrationsrate (Nierenfunktion) verwendet werden. Der Kreatinin-Serumspiegel hängt neben der Nierenfunktion auch von der Muskelmasse ab.
Biosynthese von Putrescin aus Arginin oder Ornithin
Bearbeiten| ⇓ | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
|
|
Pyridoxal- phosphat | Arginin-Decarboxylase | 4.1.1.19 | Ly | ||||||
|
||||||||||
|
Agmatinase | 3.5.3.11 | Hyd | |||||||
|
||||||||||
| CO2
|
Pyridoxal- phosphat | Ornithin-Decarboxylase | 4.1.1.17 | Ly | ||||||
|
||||||||||

Aus Putrescin werden Spermidin und Spermin gebildet
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| S-Adenosylmethioninamin | Spermidin-Synthase | 2.5.1.16 | Tr | |||||||
|
||||||||||
| S-Adenosylmethioninamin | Spermin-Synthase | 2.5.1.22 | Tr | Snyder-Robinson-S. | ||||||
|
||||||||||
Die basischen Polyamine Spermidin und Spermin interagieren mit den sauren Phosphatgruppen der DNA und regulieren das Zellwachstum. Sie kommen in allen Geweben vor, u.a. reichlich im Sperma.
Literatur
BearbeitenWeblinks
Bearbeiten- KEGG: Arginine and proline metabolism - Homo sapiens (human)
- KEGG: Urea cycle and metabolism of amino groups - Homo sapiens (human)
- RCSB PDB: Nitric Oxide Synthase
Threonin-, Glycin- und Serin-Stoffwechsel
BearbeitenAllgemeines
BearbeitenL-Serin ist eine nicht-essentielle, hydrophile, proteinogene Aminosäure. Sie wird aus einem Zwischenprodukt des Glycolyse gebildet. Benötigt wird sie für die Synthese von Glycin, Cystein, Sphingolipiden und Cholin. Außerdem liefert sie 1-Kohlenstoff-Reste in den Folsäurestoffwechsel.
Glycin ist die kleinste und einfachste Aminosäure. Sie besitzt keine Seitenkette und kann aus Serin synthetisiert werden. Verwendung findet sie in zahlreichen Biosynthesen (s.u.) und als hemmender Neurotransmitter im Rückenmark.
Threonin ist eine essentielle, polare, proteinogene Aminosäure.
Biosynthese von L-Serin aus 3-Phosphoglycerat und Deaminierung zu Pyruvat
Bearbeiten| ⇓ | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| NAD+ | NAD+ | Phosphoglycerat- Dehydrogenase | 1.1.1.95 | Ox | PHGDH-Def. (Kongenitale Mikrozephalie, psychomotorische Retardierung, Krämpfe) | |||||
|
||||||||||
| Glutamat | Glutamat | Pyridoxal- phosphat | Phosphoserin- Transaminase | 2.6.1.52 | Tr | PSAT-Def. | ||||
|
||||||||||
| H2O
Pi |
Mg2+ | Phosphoserin- Phosphatase | 3.1.3.3 | Hyd | PSP-Def. | |||||
|
||||||||||
|
|
|
Pyridoxal- phosphat | L-Serin-Deaminase | 4.3.1.17 | ||||||
|
||||||||||
Startpunkt der Serin-Biosynthese ist 3-Phosphoglycerat, ein Metabolit der Glycolyse bzw. Gluconeogenese. Serin kann zu Pyruvat deaminiert oder wie in der nächsten Tabelle dargestellt über Glycin vollständig abgebaut werden. Letzteres ist ein wichtiger Weg, um N5,N10-Methylen-Tetrahydrofolsäure zu regenerieren.
Biosynthese von Glycin aus L-Serin und Abbau
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
|
H2O, N5,N10-Methylen-THF |
N5,N10-Methylen-THF, H2O |
Pyridoxal- phosphat | Serin-Aldolase | 2.1.2.1 | Tr | |||||
|
||||||||||
| Protein-Lipoyllysin
CO2 |
Pyridoxal- phosphat | Glycin-Dehydrogenase (decarboxylierend) | 1.4.4.2 | Ox | Glycin- Enzephalopathie (GCE) | |||||
|
||||||||||
| THF
NH3, N5,N10-Methylen-THF |
Aminomethyltransferase | 2.1.2.10 | Tr | Glycin- Enzephalopathie (GCE) | ||||||
|
||||||||||
| NAD+ | FAD | Dihydrolipoyl- Dehydrogenase | 1.8.1.4 | Ox | Ahornsirup- Krankheit Typ III | |||||
|
||||||||||



Aus Serin kann der Körper Glycin bilden und vice versa. Glycin entsteht weiterhin bei der Biosynthese von Carnitin aus Protein-Lysin. Der Abbau von Glycin erfolgt entweder über Serin zu Pyruvat, bei diesem Weg muss eine Methyl(en)-Gruppe investiert werden (N5,N10-Methylen-THF) oder Glycin wird mit Hilfe eines Lipoylproteins decarboxyliert und der C1-Rest unter Ammoniakabspaltung auf THF übertragen werden, so dass im Ggs. zu vorher eine Methyl(en)-Gruppe gewonnen wird.
Abbau von L-Threonin zu Acetaldehyd und Glycin
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||||
| Pyridoxal- phosphat | Threonin-Aldolase | 4.1.2.5 | Ly | ||||||||||
|
|||||||||||||

Die essentielle Aminosäure Threonin kann auf 2 Wegen abgebaut werden. Zum einen über die Aldolspaltung zu Acetaldehyd (ketogen) und Glycin. Letzteres kann z.B. über Serin in Pyruvat (glucogen) umgewandelt werden. Die 2. Möglichkeit ist eine α-,β-Eliminierung zur Propionyl-CoA (glucogen).
Alternativer Abbau von Threonin via α-,β-Eliminierung
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
|
|
Pyridoxal- phosphat | Threonin-Ammoniak-Lyase | 4.3.1.19 | Ly | ||||||
|
||||||||||
| CoA-SH, ?
CO2, ? |
? | ? | ? | |||||||
|
||||||||||
Die Rolle der drei Aminosäuren im Stoffwechsel
BearbeitenL-Serin
Bearbeiten- Biosynthese von Glycin
- Abbau von Methionin und Biosynthese von L-Cystein
- Einschleusung von C1-Resten in den Tetrahydrofolsäure-Stoffwechsel (Bildung von N5,N10-Methylen-THF)
- Bioynthese von Phosphatidylserin
- Sphingolipid-Biosynthese
- Proteinbiosynthese
- Die Phosphorylierung zur Interkonversion von Proteinen (Konformationsänderung durch die polare Phosphatgruppe) durch spezifische Proteinkinasen (Phosphotransferasen) erfolgt meist an Aminosäuren mit einer Hydroxylgruppe (Serin > Threonin > Tyrosin).
- Serin ist neben Threonin, Hydroxyprolin und Hydroxylysin eine Verknüpfungstelle für Zucker in Glycoproteinen in Form einer O-glykosidischen Bindung.
- Die PALP-abhängige Decarboxylierung von Serin liefert das biogene Amin Ethanolamin, aus dem durch 3fache Methylierung (mittels S-Adenosylmethionin (SAM)) Cholin gebildet wird. (Anm.: Dieser Weg ist in KEGG nicht beschrieben).
Glycin
Bearbeiten- Abbau von Cholin zu Glycin
- Porphyrin-Biosynthese
- Purin-Biosynthese
- Kreatin-Biosynthese
- Glyoxylat-Stoffwechsel
- Biosynthese der Gallensäuren (Glycocholsäure)
- Einschleusung von C1-Resten in den Tetrahydrofolsäure-Stoffwechsel (Bildung von N5,N10-Methylen-THF)
- Phase II der Biotransformation in der Leber (Konjugation)
- Glutathionbiosynthese
- Proteinbiosynthese, z.B. von Kollagen (Glycin ist notwendig für die räumliche Ausbildung der Kollagen-Triplehelix)
- Im Rückenmark wirkt Glycin (ähnlich wie GABA im Gehirn) als inhibitorischer Neurotransmitter und öffnet ligandengesteuerte Chlorid-Kanäle.
L-Threonin
Bearbeiten- Proteinbiosynthese
- Phosphorylierbar (s.o.)
- O-glykosidischen Bindung in Glycoproteinen (s.o.)
Weblinks
Bearbeiten
Glyoxylat-Stoffwechsel
BearbeitenAbbau und Synthese von Glycolat und Glyoxylat
Bearbeiten| ⇓ | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| H2O
Pi |
Phosphoglycolat-Phosphatase | 3.1.3.18 | Hyd | |||||||
|
||||||||||
| NAD(P)+
NAD(P)H/H+ |
NAD(P)+
NAD(P)H/H+ |
Glyoxylat-Reduktase (NAD+) | 1.1.1.26 | Ox | ||||||
| Glyoxylat-Reduktase/ Hydroxypyruvat-Reduktase (NADP+) | 1.1.1.79 | Ox | Primäre Hyperoxalurie (Oxalose) II | |||||||
|
H2O2 |
oder |
FMN | Glycolat-Oxidase | 1.1.3.15 | Ox | |||||
|
||||||||||
| L-Alanin | L-Alanin | Pyridoxal- phosphat | Alanin--Glyoxylat-Transaminase | 2.6.1.44 | Tr | Primäre Hyperoxalurie (Oxalose) I | ||||
|
||||||||||

Pathobiochemie
BearbeitenBei der primären Hyperoxalurie Typ I (Oxalose Typ I) lagert sich Oxalat in verschiedenen Geweben ab. In den Harnwegen bilden sich Oxalatsteine.
Weblinks
Bearbeiten
Lysin-Stoffwechsel
BearbeitenAllgemeines
BearbeitenL-Lysin ist eine essentielle, proteinogene Aminosäure mit einer basischen, positiv geladenen Seitenkette. Im Kollagen kommt es als Hydroxylysin vor (posttranslationale Modifikation). Lysin ist weiterhin der Ausgangspunkt für die Biosynthese von Carnitin.
Abbau der essentiellen Aminosäure L-Lysin zu 2 Acetyl-CoA
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| α-Ketoglutarat, NAD(P)H/H+
|
|
Saccharopin-Dehydrogenase | 1.5.1.7 | Ox | Saccharopinurie, Hyperlysinämie | |||||
| Saccharopin-Dehydrogenase | 1.5.1.8 | |||||||||
|
||||||||||
| NAD(P)+, H2O | H2O, NAD(P)+ | Saccharopin-Dehydrogenase | 1.5.1.9 | Ox | Hyperlysinämie | |||||
| Saccharopin-Dehydrogenase | 1.5.1.10 | |||||||||
|
||||||||||
| H2O, NAD+ | Aminoadipat-semialdehyd-Dehydrogenase | 1.2.1.31 | Ox | |||||||
|
||||||||||
| α-Ketoglutarat | α-Ketoglutarat | Pyridoxal- phosphat | α-Aminoadipat-Transaminase | 2.6.1.39 | Tr | |||||
|
||||||||||
| CoA-SH, NAD+
CO2, NADH/H+ |
Thiamin-P2 | α-Ketoglutarat-Dehydrogenase-Komplex | 1.2.4.2 | Ox | α-Ketoglutarat-Dehydrogenase-Defizienz | |||||
| Tr | ||||||||||
| FAD | 1.8.1.4 | Ox | MSUD III | |||||||
|
||||||||||
| FAD
CO2, FADH2 |
FAD | Glutaryl-CoA-Dehydrogenase | 1.3.99.7 | Ox | Glutaracidurie Typ I | |||||
|
||||||||||
| H2O
|
Enoyl-CoA-Hydratase Mitochondrium | 4.2.1.17 | Ly | TFP-Defizienz | ||||||
|
||||||||||
| NAD+ | NAD+ | β-Hydroxyacyl-CoA-Dehydrogenase | 1.1.1.35 | Ox | HADH-Defizienz | |||||
|
||||||||||
| CoA-SH
|
|
CoA-SH
|
Acetyl-CoA-C-Acyltransferase (β-Ketothiolase) | 2.3.1.16 | Tr | TFP-Defizienz | ||||
| Acetyl-CoA-C-Acetyltransferase (Acetoacetyl-CoA-Thiolase) | 2.3.1.9 | α-Methylacetoacetacidurie | ||||||||
|
||||||||||




Da für die Abspaltung der ε-Aminogruppe des Lysins kein eigenes Enzym zur Verfügung steht, bedient sich die Zelle eines anderen Mechanismus, den wir vom Aspartatzyklus her schon kennen. Lysin wird dabei mit einem Akzeptor-Molekül (hier α-Ketoglutarat) kondensiert und dann wieder unter Übertragung der Aminogruppe (α-Ketoglutarat wird zu L-Glutamat) abgespalten.
Anschließend erfolgt die Dehydrierung zur Monoaminodicarbonsäure, die zweite Transaminierung, eine dehydrierende Decarboxylierung (Aktivierung mit CoA) und der weitere Abbau über Teilschritte der β-Oxidation (hier mit einer Decarboxylierung) bis zu den zwei Acetyl-CoA, die in den Citratzyklus eingeschleust werden können.
α-Ketoadipat fällt auch beim Tryptophan-Abbau an, so dass ab hier eine gemeinsame Endstrecke besteht.
Biologische Funktion
BearbeitenWie alle proteinogenen Aminosäuren ist Lysin Bestandteil der Proteine. Im Protein kann es für positive Ladungen sorgen (Protonenanlagerung an die basische ε-Aminogruppe bei zellulärem pH). Im Kollagen kommt es in hydroxylierter Form vor. Im Kollagen und Fibrin hilft es bei der Quervernetzung der Proteinmonomere. Weiterhin ist es Ausgangspunkt der Carnitin-Biosynthese.
Weblinks
Bearbeiten
Carnitin-Stoffwechsel
BearbeitenAllgemeines
BearbeitenCarnitin wird aus der Aminosäure Lysin gebildet. Es dient als Transportmittel für Fettsäuren über die innere Mitochondrienmembran.
Biosynthese von Carnitin aus Lysin
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| S-Adenosylmethionin | Protein-Lysin-N-Methyltransferase | 2.1.1.43 | Tr | |||||||
|
||||||||||
| S-Adenosylmethionin | Protein-Lysin-N-Methyltransferase | 2.1.1.43 | Tr | |||||||
|
||||||||||
| S-Adenosylmethionin | Protein-Lysin-N-Methyltransferase | 2.1.1.43 | Tr | |||||||
|
||||||||||
|
|
Peptid-Hydrolase | 3.4.-.- | Hyd | |||||||
|
||||||||||
| α-Ketoglutarat, O2
Succinat, CO2 |
Fe, Ascorbat | Trimethyllysin-Dioxygenase | 1.14.11.8 | Ox | ||||||
|
||||||||||
| Pyridoxal- phosphat | Glycin-Hydroxymethyltransferase | 2.1.2.1 | Tr | |||||||
|
||||||||||
| H2O, NAD+ | Aldehyd-Dehydrogenase (NAD+) | 1.2.1.3 | Ox | ALDH2-Def., Sjogren-Larsson-S. | ||||||
| 4-Trimethylammoniobutyraldehyd- Dehydrogenase | 1.2.1.47 | |||||||||
|
||||||||||
| α-Ketoglutarat, O2
Succinat, CO2 |
Fe, Ascorbat | γ-Butyrobetain-Dioxygenase | 1.14.11.1 | Ox | ||||||
|
||||||||||







Proteingebundens Lysin wird drei mal methyliert, wobei die Methylgruppe von S-Adenosylmethionin (SAM) stammt. Das Trimethyllysin wird dann durch eine Hydrolase abgespalten und Vitamin C-abhängig zum Hydroxy-Trimethyllysin hydroxyliert. Durch Abspaltung eines Glycins entsteht 4-Trimethylammoniobutanal. Dieses wird dann oxidiert und ein zweites Mal Vitamin C-abhängig hydroxyliert, so dass Carnitin entsteht.
Biologische Funktion
BearbeitenCarnitin dient als Carriermolekül für langkettige Fettsäuren, die erst an das Trägermolekül gekoppelt die innere Mitochondrienmembran überwinden können. Siehe unter β-Oxidation.
Pathobiochemie
BearbeitenMögliche Defekte:
- Carnitin-O-Palmitoyltransferase-Defizienz: CPT1A-Defizienz, CPT1A-Defizienz
- Carnitin-Acylcarnitin-Translocase-Defizienz (CACT-Defizienz)
- Systemische primäre Carnitin-Defizienz - Defekt im Gen SLC22A5 (5q31.1), das für den sodium ion-dependent carnitine transporter OCTN2 kodiert. OMIM
Eine Defizienz des Carnitin-Systems führt zu einer Beeinträchtigung der beta-Oxidation.
Literatur
Bearbeiten- Longo N, Amat di San Filippo C, Pasquali M. “Disorders of carnitine transport and the carnitine cycle”. Am J Med Genet C Semin Med Genet, 142C:77–85, May 2006. DOI:10.1002/ajmg.c.30087. PMID 16602102.
- Stanley CA. “Carnitine deficiency disorders in children”. Ann. N. Y. Acad. Sci., 1033:42–51, November 2004. DOI:10.1196/annals.1320.004. PMID 15591002.
Weblinks
Bearbeiten
Methionin-Stoffwechsel
BearbeitenAllgemeines
BearbeitenL-Methionin ist eine essentielle, schwefelhaltige proteinogene Aminosäure. Das Methionin-Derivat S-Adenosylmethionin dient an vielen Stellen des Stoffwechsels als Methylgruppen-Lieferant. Weiterhin ist die Bildung der Aminosäure Cystein aus Serin mit dem Methionin-Abbau verknüpft. Die Rückgewinnung von Methionin aus Homocystein benötigt Cobalamin und Folsäure (5-Methyl-THF), dabei wird 5-Methyl-THF, die Transportform der Folsäure im Blut, in die biologisch vielseitiger einsetzbare Tetrahydrofolsäure (THF) überführt.
== Abbau von Homocystein
==
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| ATP
3 Pi |
Methionin- Adenosyltransferase | 2.5.1.6 | Tr | Hypermeth- ioninämie | ||||||
|
||||||||||
| Akzeptor
Akzeptor-CH3 |
Akzeptor
Akzeptor-CH3 |
Bsp.:
DNA(Cytosin-5-)- Methyltransferase |
2.1.1.37 | Tr | ICF-S. | |||||
|
||||||||||
| H2O | H2O | NAD | Adenosylhomocysteinase | 3.3.1.1 | Hyd | Hypermeth- ioninämie | ||||
|
||||||||||
|
Pyridoxal- phosphat | Cystathionin-β-Synthase | 4.2.1.22 | Ly | Homo- cystinurie I | |||||
|
||||||||||
|
Pyridoxal- phosphat | Cystathionin-γ-Lyase | 4.4.1.1 | Ly | Cystathio- ninurie | |||||
|
||||||||||
| CoA-SH, ?
CO2, ? |
NAD | α-Ketosäure-Decarboxylase | ? | ? | ||||||
|
||||||||||



Biologische Rolle
BearbeitenDas schwefelhaltige L-Methionin ist eine für den Menschen und viele Tiere essentielle Aminosäure. Der Abbauweg von Methionin hat mehrere Bedeutungen:
- Produktion des Methylgruppen-Donors S-Adenosylmethionin (SAM).
- Bildung der proteinogenen schwefelhaltigen Aminosäure L-Cystein aus L-Serin (L-Cystein ist für Säuglinge eine essentielle Aminosäure, da bei ihnen die entsprechende Enzymausstattung noch nicht ausgereift ist.)
- Abbau des Restes zu Propionyl-CoA.
Funktion von S-Adenosylmethionin als Methylgruppen-Donor
Bearbeiten- Im Stoffwechsel der aromatischen Aminosäuren bzw. der biogenen Amine:
- Methylierung von DNA-Basen, z.B. von Cytosin zu 5-Methylcytosin
- Methylierung von Guanidinoacetat zu Kreatin
- Biosynthese von Carnitin aus Lysin
- Methylierung von Ethanolamin zu Cholin
- Methylierung von Glycin zu Sarcosin (wahrscheinlich zur Senkung des S-Adenosylmethionin-Angebots)
- Biosynthese von Ubichinon
- Methylierung von Cobalamin zu Methylcobalamin zur Verwendung an der Methionin-Synthase (reduktive Methylierung)
Rückgewinnung von Methionin aus Homocystein
Bearbeiten| (⇓) | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| THF | Methylcobalamin, Zn | Methionin-Synthase | 2.1.1.13 | Tr | Methylcobalamin-Def., Typ cblG | |||||
| ferner:
|
Zn | ferner:
Betain--Homocystein- S-Methyltransferase |
2.1.1.5 | Tr | ||||||
|
||||||||||
Da u.U. mehr Methyl-Gruppen bzw. S-Adenosylmethionin benötigt wird, als Methionin abgebaut wird, kann Methionin durch Methylierung von Homocystein regeneriert werden. Die Methylgruppe stammt dabei entweder aus dem Folsäure-Stoffwechsel (also meist aus dem Serin- und Glycin-Abbau) oder aus dem Cholin-Abbau (Betain). Die mit Folat arbeitende Methionin-Synthase benötigt Cobalamin (Vitamin B12) als Cofaktor. Die 2. Funktion dieses Stoffwechselschritts besteht darin, die Transportform der Folsäure 5-Methyl-THF in die biologisch aktivere Tetrahydrofolsäure (THF) umzuwandeln.
Die Methionin-Synthase ist auf reduziertes (Methyl-)Cob(I)alamin als Kofaktor angewiesen. Dieses wird im Laufe der Zeit jedoch zu Cob(II)alamin oxidiert und damit die Enzymaktivität herabgesetzt. Reaktiviert wird das Enzym bzw. der Kofaktor durch eine reduktive Methylierung an der Methionin-Synthase-Reduktase mit S-Adenosylmethionin (SAM) als Methylgruppen-Donor.
Bildung von 5'-Methylthioadenosin
BearbeitenIm Rahmen der Polyaminbiosynthese wird S-Adenosylmethioninamin benötigt, um die basischen Polyamine Spermidin und Spermin mit Aminopropyl-Resten zu verlängern. Es entsteht 5'-Methylthioadenosin, das im Folgenden wieder zu Methionin recycliert werden kann.
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
|
|
Pyruvat | Adenosylmethionin- Decarboxylase | 4.1.1.50 | Ly | ||||||
|
||||||||||
| Putrescin/Spermidin | Spermidin-Synthase | 2.5.1.16 | Tr | |||||||
| Spermin-Synthase | 2.5.1.22 | Tr | Snyder-Robinson-S. | |||||||
|
||||||||||


Methionin-Salvage
BearbeitenDer Salvage-(Rückgewinnungs-)Weg für Methionin verhindert, dass Schwefel energieaufwändig neu assimiliert werden muss. Die auf die Polyamine übertragene Aminopropylgruppe wird in sechs Reaktionsschritten wieder aus dem Ribosering hergestellt.
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| Pi | S-Methyl-5-thioadenosin- Phosphorylase | 2.4.2.28 | Tr | |||||||
|
||||||||||
| 5′-Methylthioribose-1-phosphat-Isomerase | 5.3.1.23 | Iso | ||||||||
|
||||||||||
|
H2O |
5′-Methylthioribulose-1-phosphat-Dehydratase | 4.2.1.109 | Ly | |||||||
|
||||||||||
| H2O
Pi, 2H+ |
Enolase-Phosphatase E1 | 3.1.3.77 | Hy | |||||||
|
||||||||||
| O2
Formiat |
Acireducton-Dioxygenase | 1.13.11.54 | Ox | |||||||
|
||||||||||
| R-CH(NH3+)-COO−
R-CO-COO− |
(Transaminase) | 2.6.1.- | Tr | |||||||
|
||||||||||


-2,3-dioxopentyl_phosphate.svg)


Weblinks
Bearbeiten- KEGG: Cysteine and methionine metabolism - Homo sapiens (human)
- KEGG: Propanoate metabolism - Homo sapiens (human)
Cystein-Stoffwechsel
BearbeitenAllgemeines
Bearbeiten
L-Cystein ist eine nicht-essentielle, Schwefel-haltige, hydrophile, proteinogene Aminosäure.
Biosynthese und Abbau
BearbeitenL-Cystein ist außer für Feten und Neugeborene keine essentielle Aminosäure, da sie im Abbauweg des Methionins aus L-Serin gebildet werden kann. Cystein kann zu Pyruvat abgebaut werden.
Biologische Bedeutung
Bearbeiten- Eine wichtige Rolle spielt die SH-Gruppe von Cystein in Proteinen, wo zwei Cystein-Reste durch Oxidation eine Disulfidbrücke (S-S) ausbilden können. Zwei über eine Disulfidbrücke verbundene Cysteine nennt man Cystin.
- Als Bestandteil des Glutathions ist die SH-Gruppe von Cystein für das Einfangen radikaler Sauerstoffspezies (ROS) z.B. in Erythrozyten zuständig.
- Weiterhin ist Cystein Ausgangsstoff des Taurins, ein Bestandteil der Gallensäure Taurocholat.
- Cystein bzw. dessen biogenes Amin Cysteamin ist ein Bestandteil von Coenzym A.
- Cystein liefert den Schwefel für die Bildung des Molybdän-Cofaktors, dabei wird L-Alanin frei.
Weblinks
Bearbeiten
Taurin- und Hypotaurin-Stoffwechsel
BearbeitenAllgemeines
BearbeitenTaurin wird für die Bildung der Gallensäure Taurocholat benötigt.
Biosynthese von Hypotaurin aus Cystein
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| O2
|
Fe, NAD(P)H | Cystein-Dioxygenase | 1.13.11.20 | Ox | ||||||
|
||||||||||
|
|
Pyridoxal- phosphat | Glutamat-Decarboxylase 1 | 4.1.1.15 | Ly | GAD1-Def. | |||||
| Sulfinoalanin-Decarboxylase | 4.1.1.29 | |||||||||
|
||||||||||

Biosynthese von Taurin aus 3-Sulfino-L-Alanin und Bildung von Taurocholsäure
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| H2O, NAD+ | Cysteinsulfinsäure-Dehydrogenase | ? | ||||||||
|
||||||||||
|
|
Pyridoxal- phosphat | Glutamat-Decarboxylase 1 | 4.1.1.15 | Ly | GAD1-Def. | |||||
| Sulfinoalanin-Decarboxylase | 4.1.1.29 | |||||||||
|
||||||||||
| Gallensäure-CoA | Gallensäure-CoA:Aminosäure-N-Acyltransferase | 2.3.1.65 | Tr | |||||||
|
||||||||||

Biologische Aufgaben
BearbeitenDas Amin Taurin ist ein Bestandteil der Gallensäure Taurocholsäure.
Weblinks
Bearbeiten
Phenylalanin-Stoffwechsel
BearbeitenAllgemeines
BearbeitenPhenylalanin ist eine essentielle proteinogene, aromatische, hydrophobe Aminosäure. Sie kann zur Proteinbiosynthese und zur Biosynthese von Tyrosin verwendet werden.
Biosynthese von Tyrosin aus Phenylalanin
Bearbeiten| Kov. | All. | Koop. | ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||
| + Phosph. | - Trp, Tyr
+ Lysolecithin, Phospholipide |
+ Phe | Tetrahydrobiopterin, O2 | Fe |
|
1.14.16.1 | Ox | Phenyl- ketonurie (PKU) | ||||


Die Phenylalanin-Hydroxylase katalysiert die Hydroxylierung der essentiellen proteinogenen Aminosäure L-Phenylalanin zu L-Tyrosin unter Beteiligung von Tetrahydrobiopterin (BH4).
Pathobiochemie: Aus einem angeborenen Defekt der Phenylalaninhydroxylase resultiert das Krankheitsbild der klassischen Phenylketonurie. Die vermehrt gebildeten z.T. toxischen Alternativ-Metaboliten führen nach der Geburt unbehandelt zu einer schweren Beeinträchtigung der Gehirnentwicklung. Die verminderte Tyrosin-Bildung kann bei fehlender Zufuhr über die Nahrung zu einem Mangel an Schilddrüsenhormon, Melanin und Katecholaminen führen. Tyrosin wird hier zur essentiellen Aminosäure. Die Behandlung der PKU ist diätetisch. Das Diätregime wird im Erwachsenenalter bzw. nach Abschluß der Hirnentwicklung oft etwas weniger eng gehandhabt, allerdings folgt aus einer ungeplanten Schwangerschaft der PKU-betroffenen Frau bei nicht eingehaltener Diät die Schädigung des ungeborenen Kindes. Weitaus seltenere Störungen der Reaktion an der Phenylalanin-Hydroxylase (und an anderen Hydroxylasen, die aromatische Aminosäurenseitenketten hydroxylieren) können durch einen Mangel des Cofaktors Tetrahydrobiopterin bedingt sein, durch Defekte im Biopterin-Stoffwechsel (siehe BH4-defiziente Phenylketonurie).
Alternative Abbauwege des Phenylalanins
BearbeitenAbbau zu Phenylenolpyruvat bzw. 2-Hydroxyphenylacetat
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||
| α-Ketoglutarat | α-Ketoglutarat | Pyridoxal- phosphat | Aspartat-Transaminase (ASAT, GOT) | 2.6.1.1 | Tr | ||||||
| Tyrosin-Transaminase | 2.6.1.5 | Tyrosinämie II (Richner-Hanhart-S.) | |||||||||
|
Phenylpyruvat-Tautomerase | 5.3.2.1 | Iso | ||||||||
| O2
CO2 |
Fe | 4-Hydroxyphenylpyruvat- Dioxygenase | 1.13.11.27 | Ox | Tyrosinämie III, Hawkinsinurie | ||||||
|
|||||||||||



Phenylalanin verliert seine Aminogruppe durch pyridoxalabhängige Transaminierung und wird anschließend zu 2-Hydroxyphenylacetat decarboxyliert.
Dieser Abbauweg entspricht den ersten zwei Schritten des Tyrosinabbaus.
Abbau zu 2-Phenylacetat
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
|
|||||||
|
CO2 |
Pyridoxal- phosphat | DOPA-Decarboxylase | 4.1.1.28 | Ly | AADC-Defizienz | ||
|
|||||||
| H2O, O2
NH3, H2O2 |
FAD | Monoamino-Oxidase (MAO) | 1.4.3.4 | Ox | MAO A: Brunner-Syndrom, Antisozialität | ||
| Cu, TPQ | Diamino-Oxidase (DAO) | 1.4.3.6 | |||||
|
|||||||
| H2O, NAD(P)+ | H2O, NAD(P)+ | Aldehyd-Dehydrogenase (ALDH) (NAD(P)+) | 1.2.1.5 | Ox | |||
|
|||||||

Phenylalanin wird bei dieser Variante zuerst decarboxyliert und dann oxidativ desaminiert. Danach erfolgt die Oxidation zu 2-Phenylacetat.
Dieser Abbauweg entspricht dem alternativen Abbau von Tyrosin zu 4-Hydroxyphenylacetat.
Abbau zu 2-Phenylacetamid
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
|
|||||||
| H2O2
CO2, 2 H2O |
|
Häm | Lactoperoxidase | 1.11.1.7 | Ox | ||
|
|||||||

Weblinks
Bearbeiten- KEGG: Phenylalanine, tyrosine and tryptophan biosynthesis - Homo sapiens (human)
- KEGG: Phenylalanine metabolism - Homo sapiens (human)
Tyrosin-Stoffwechsel
BearbeitenAllgemeines
BearbeitenTyrosin (tyros (griech): Käse) ist eine proteinogene aromatische Aminosäure. Sie ist nicht essentiell, da sie aus Phenylalanin synthetisiert werden kann. Aus Tyrosin gehen die Schilddrüsenhormone, die Katecholamine und das Melanin der Haut hervor. In Proteinen kann die OH-Gruppe von Tyrosin (wie bei Serin und Threonin) phosporyliert werden.
Abbau von Tyrosin zu Acetoacetat und Fumarat
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||||
| α-Ketoglutarat | α-Ketoglutarat | Pyridoxal- phosphat | Aspartat-Transaminase (Aspartat-Aminotransferase) (AST, ASAT, GOT) | 2.6.1.1 | Tr | ||||||||
| Tyrosin-Transaminase Leber | 2.6.1.5 | Tyrosinämie II (Richner-Hanhart-S.) | |||||||||||
|
Phenylpyruvat-Tautomerase | 5.3.2.1 | Iso | ||||||||||
| O2
CO2 |
Fe | 4-Hydroxyphenylpyruvat-Dioxygenase | 1.13.11.27 | Ox | Tyrosinämie III, Hawkinsinurie | ||||||||
|
|||||||||||||
| O2
|
Fe | Homogentisat-1,2-Dioxygenase | 1.13.11.5 | Ox | Alkaptonurie | ||||||||
|
|||||||||||||
| Maleylacetoacetat-Isomerase | 5.2.1.2 | Iso | |||||||||||
|
|||||||||||||
| H2O
|
Fumarylacetoacetase | 3.7.1.2 | Hyd | Tyrosinämie I | |||||||||
|
|||||||||||||



Die ersten beiden Schritte entsprechen dem alternativen Abbau von Phenylalanin zu 2-Hydroxyphenylacetat.
Acetoacetat ist ein Ketonkörper, Fumarat ist ein Intermediat des Citratzyklus.
Alternativer Abbauweg von Tyrosin zu Tyramin und 4-Hydroxyphenylacetat
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
|
|
Pyridoxal- phosphat | DOPA-Decarboxylase | 4.1.1.28 | Ly | AADC-Defizienz | |||||
|
||||||||||
| H2O, O2
NH3, H2O2 |
FAD | Monoamino-Oxidase (MAO) | 1.4.3.4 | Ox | MAO A: Brunner-Syndrom, Antisozialität | |||||
| Cu, TPQ | Diamino-Oxidase (DAO) | 1.4.3.6 | ||||||||
|
||||||||||
| H2O, NAD(P)+ | NAD(P)+, H2O | Aldehyd-Dehydrogenase (ALDH) (NAD(P)+) | 1.2.1.5 | Ox | ||||||
|
||||||||||



Diese Abbauschritte inklusive dem Enzym MAO-A finden Sie auch im alternativen Abbau von Phenylalanin zu Phenylacetat und eingestreut im Abbauweg der Katecholamine (s.u.).
Tyramin kann auch in Dopamin umgewandelt werden:
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
|
|||||||
| O2, NADH/H+
H2O, NAD+ |
Cu | Tyrosinase (Monophenol-Monooxygenase) | 1.14.18.1 | Ox | Okulokutaner Albinismus IA, Okulokutaner Albinismus IB | ||
|
|||||||

Pharmakologie: Reversible MAO-A-Hemmer wie Moclobemid wirken durch Steigerung der Monoaminkonzentrationen (Abbauhemmung v.a.von Noradrenalin und Serotonin) im ZNS antidepressiv und antriebssteigernd. Unspezifische irreversible MAO-Hemmer wie Tranylcypromin hemmen auch verstärkt den Abbau von Tyramin, so dass Tyramin-reiche Lebensmittel wie Käse, Bananen und Schokolade zur Tyraminakkumulation führen können. Dieses wird dann vermehrt in Dopamin umgewandelt und führt zu sympathikotonen Blutdruckkrisen („Cheese-reaction“).
Biosynthese von Triiodthyronin (T3) und L-Thyroxin (T4)
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
|
|||||||
| Iodid, H2O2
2 H2O |
Häm | Thyreoperoxidase (TPO) Schilddrüse | 1.11.1.8 | Ox | Thyroiddyshormonogenesis 2A | ||
|
|||||||
| Iodid, H2O2
2 H2O |
Häm | Thyreoperoxidase (TPO) Schilddrüse | 1.11.1.8 | Ox | Thyroiddyshormonogenesis 2A | ||
|
|||||||




Die zum Thyreoglobulin gehörenden Tyrosylreste werden zuerst von der Thyreoperoxidase zum Mono- oder Diiodtyrosin jodiert. Diese Reaktion findet an den Mikrovilli am apikalen Teil der Plasmamembran der Schilddrüsenepithelzellen statt. Eines dieser Moleküle wird dann auf ein anderes übertragen, so dass Triiodthyronin (T3) oder Tetraiodthyronin (L-Thyroxin, T4) entsteht. Das Thyreoglobulin mit den T3- und T4-Resten wird extrazellulär in den Schilddrüsenfollikeln (Kolloid) gespeichert. Die Schilddrüse sezerniert zum größeren Teil das biologisch weniger aktive T4 (HWZ: 7 Tage, höhere Bindung an das Thyroxin-bindende Globulin (TBG)) und zum kleineren Teil das wirksamere T3 (HWZ: 1 Tag). Peripher wird ein Teil des T4 von der Thyroxin-5'-Deiodinase (EC 1.97.1.10) in T3 umgewandelt. Biologisch aktiv ist wie bei allen Hormonen nur der freie, nicht-proteingebundene Anteil, d.h. hier fT3 und fT4.
Die Schilddrüsenhormone können von der Thyroxin-5'-Deiodinase (EC 1.97.1.11) inaktiviert werden oder sie werden in der Leber mit Glucuronat oder Sulfat konjugiert und mit der Galle ausgeschieden. Eine dritte Möglichkeit besteht in der oxidativen Deaminierung und Decarboxylierung der Alanylseitenkette, so dass Tri- und Tetrajodthyreoacetat entsteht, das nach Deiodierung ausgeschieden wird.
Stimuliert wird die Hormonsekretion durch TSH, das von der Adenohypophyse ins Blut sezerniert wird, gesteuert vom TRH des Hypothalamus.
Pathophysiologie: Eine Hyperthyreose supprimiert den Regelkreis und die TSH-Bildung, eine Hypothyreose stimuliert ihn. Daher ist der TSH-Spiegel (neben fT3 und fT4) ein wichtiger Parameter in der Schilddrüsendiagnostik.
Pharmakologie: Bei der Behandlung der Hypothyreose substituiert man üblicherweise mit T4, da dieses die günstigere Pharmakokinetik aufweist (lange Halbwertszeit) und vom Körper nach Bedarf zu T3 umgesetzt wird.
Biosynthese von Melanin
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | ||
|---|---|---|---|---|---|---|---|---|---|
|
|||||||||
|
Cu | Tyrosinase (Monophenol-Monooxygenase) | 1.14.18.1 | Ox | Okulokutaner Albinismus IA, Okulokutaner Albinismus IB | ||||
|
|||||||||
| Ox. Akzeptor
Red. Akzeptor |
|||||||||
|
|||||||||
| Dopachinon
L-Dopa |
|||||||||
|
|||||||||
|
|
Cu | Tyrosinase (Monophenol-Monooxygenase) | 1.14.18.1 | Ox | Okulokutaner Albinismus IA, Okulokutaner Albinismus IB | ||||
|
|||||||||
| O2, DHI
2 H2O, Indol-5,6-chinon |
Cu | Tyrosinase (Monophenol-Monooxygenase) | 1.14.18.1 | Ox | Okulokutaner Albinismus IA, Okulokutaner Albinismus IB | ||||
|
|||||||||
| O2
H2O |
|||||||||
|
|||||||||






2. Möglichkeit von Dopachrom ausgehend:
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| Zn | L-Dopachrom-Isomerase | 5.3.3.12 | Iso | |||||||
|
||||||||||
| 2 Ox. Akz.
2 Red. Akz., CO2 |
tyrosinase-related protein 1 | 1.14.18.- | Ox | |||||||
|
||||||||||

Melanin wird u.a. von den Melanozyten der Haut synthetisiert und über Zellfortsätze an die Keratinozyten abgegeben. Es schützt die Haut vor UV-Licht. Auch die Iris enthält Melanin.
Pathobiochemie: Melanin-Biosynthesestörungen z.B. bei einem Tyrosinase-Mangel führen zum Albinismus.
Biosynthese der Katecholamine
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| Tetrahydrobiopterin, O2 | Fe | Tyrosin-Hydroxylase (Tyrosin-3-Monooxygenase) | 1.14.16.2 | Ox | Segawa-Syndrom | |||||
|
||||||||||
|
|
Pyridoxal- phosphat | DOPA-Decarboxylase | 4.1.1.28 | Ly | AADC-Defizienz | |||||
|
||||||||||
| Ascorbat, O2
Dehydroascorbat, H2O |
Cu | Dopamin-β-Monooxygenase (Dopamin-β-Hydroxylase) | 1.14.17.1 | Ox | DBH-Defizienz | |||||
|
||||||||||
| S-Adenosylmethionin | Noradrenalin-N-Methyltransferase | 2.1.1.28 | Tr | |||||||
|
||||||||||

 |
Dopamin ist ein wichtiger Neurotransmitter im ZNS. Noradrenalin (Norepinephrin) spielt als Neurotransmitter im ZNS und an den sympathischen Nervenenden eine große Rolle. Adrenalin (Epinephrin) wird v.a. im Nebennierenmark gebildet und bei Stress ins Blut sezerniert.
Pathophysiologie und Pharmakologie: Dopaminmangel im Corpus striatum durch Untergang dopaminerger Neurone in der Substantia nigra führt zum Morbus Parkinson. Der progrediente Dopaminmangel kann u.a. durch Gabe von L-DOPA (,das im Ggs. zum Dopamin die Blut-Hirn-Schranke passieren kann) günstig beeinflusst werden. L-DOPA-Präparate enthalten fast immer einen DOPA-Decarboxylase-Hemmer (z.B. Carbidopa oder Benserazid) in fixer Kombination. Dieser kann die Blut-Hirn-Schranke nicht überwinden und verhindert in der Peripherie, dass L-DOPA zum Dopamin decarboxyliert wird und damit im ZNS nicht mehr ankommt. Dadurch können die L-DOPA-Dosis reduziert und periphere dopaminerge Nebenwirkungen vermindert werden.
Adrenalin wirkt v.a. auf das Herz und wird in der Reanimation und beim anaphylaktischen Schock eingesetzt, Noradrenalin wirkt v.a. vasokonstriktorisch und bietet sich z.B. zur Therapie des (nicht-kardiogenen) Schocks an. Der kardiogene Schock wird mit dem synthetischen Katecholamin Dobutamin behandelt.
Abbau der Katecholamine
Bearbeiten| ⇓ | Subst. | Enz./EG | Erkr. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
 Noradrenalin Noradrenalin
|
 Adrenalin Adrenalin
|
Noradrenalin
|
Adrenalin
|
||||||||
| SAM | COMT 2.1.1.6 Tr | ||||||||||
 L-Metanephrin L-Metanephrin
|
|||||||||||
| H2O, O2
H2O2, NH3/ Methylamin |
MAO (Co: FAD) 1.4.3.4 Ox | Brunner-Syndrom, Antisozialität | |||||||||
| H2O, NAD(P)+ | ALDH (NAD(P)+) 1.2.1.5 Ox | ||||||||||
 3,4-Dihydroxy- mandelat 3,4-Dihydroxy- mandelat
|
|||||||||||
| NAD(P)H/H+ | ADH (Co: Zn/Fe)
1.1.1.1 Ox |
||||||||||
 3,4-Dihydroxy- phenylethylenglycol 3,4-Dihydroxy- phenylethylenglycol
|
|||||||||||
| SAM | COMT 2.1.1.6 Tr | ||||||||||
|
|||||||||||

 |
 |
 |
 |
Für den Abbau der Katecholamine sind besonders zwei Enzyme von Bedeutung: Die in der äußeren Mitochondrienmembran lokalisierte Monoamino-Oxidase (MAO) und die Catechol-O-Methyltransferase (COMT). Daneben sind die Aldehyd-Dehydrogenase (ALDH) und die Alkohol-Dehydrogenase (ADH) am Abbau beteiligt.
Aus der Tabelle werden 3 Abbauwege ersichtlich:
a) Dopamin, Noradrenlin und Adrenalin: 1. Methylierung, 2. Oxidative Deaminierung zum Aldehyd, 3. Oxidation zur Säure.
b) Dopamin, Noradrenlin und Adrenalin: 1. Oxidative Deaminierung zum Aldehyd, 2. Oxidation zur Säure, 3. Methylierung.
c) Noradrenalin und Adrenalin: 1. Oxidative Deaminierung zum Aldehyd, 2. Reduktion zum Alkohol, 3. Methylierung.
a) und b) unterscheiden sich nur in der Reihenfolge. In beiden Fällen resultiert aus der Oxidation des Aldehyds (CHO) eine Säure (COOH), in c) ergibt sich aus der Reduktion ein Alkohol (OH).
Der Dopaminabbau führt über a) oder b) zum Homovanillat, der Abbau von Noradrenalin und Adrenalin führt über a) oder b) zur Vanilinmandelsäure oder über c) zum 3-Methoxy-4-hydroxy-phenylethylenglycol.
Klinik: Der Verdacht auf ein Phäochromozytom, ein Katecholaminproduzierender Tumor des Nebennierenmarks, lässt sich durch Bestimmung der Metanephrine im Serum (erhöht), ggf. auch durch Nachweis einer gesteigerten Vanilinmandelsäureausscheidung im 24-Stunden-Urin erhärten.
Pharmakologie: Der Katecholaminabbau kann mit MAO- und COMT-Hemmern gebremst werden. Therapeutisch eingesetzt werden COMT-Hemmer (z.B. Entacapon) und MAO-B-Hemmer (z.B. Selegilin) beim Morbus Parkinson. MAO-A-Hemmer (z.B. Moclobemid) und unspezifische MAO-Hemmer (z.B. Tranylcypromin) wirken antidepressiv und antriebssteigernd, indem sie das Transmitterangebot an den Synapsen erhöhen.
Weblinks
Bearbeiten
Tryptophan-Stoffwechsel
BearbeitenAllgemeines
BearbeitenTryptophan ist eine essentielle, proteinogene Aminosäure. Die Seitenkette enthält ein lipophiles aromatisches Indolringsystem. Tryptophan ist der Rohstoff der biogenen Amine Tryptamin, 5-Hydroxytryptamin (Serotonin) und Melatonin. Des weiteren zweigt die Biosynthese von Nikotinsäure (Vitamin B3) vom Tryptophan-Stoffwechsel ab.
Decarboxylierung von Tryptophan zu Tryptamin und Abbau
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
|
|
Pyridoxal- phosphat | DOPA-Decarboxylase | 4.1.1.28 | Ly | AADC-Def. | |||||
|
||||||||||
| H2O, O2
H2O2, NH3 |
FAD | Monoamino-Oxidase (MAO) | 1.4.3.4 | Ox | MAO A: Brunner-S., Antisozialität | |||||
| Cu, TPQ | Diamino-Oxidase (DAO) | 1.4.3.6 | Ox | |||||||
|
||||||||||
| H2O, NAD+ | Aldehyd-Dehydrogenase (NAD+) | 1.2.1.3 | Ox | |||||||
|
||||||||||




Biosynthese von Serotonin aus L-Tryptophan und Abbau
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| Tetrahydrobiopterin, O2 | Fe | Tryptophan-Hydroxylase (Tryptophan-5-Monooxygenase) | 1.14.16.4 | Ox | ||||||
|
||||||||||
|
|
Pyridoxal- phosphat | DOPA-Decarboxylase | 4.1.1.28 | Ly | AADC-Def. | |||||
|
||||||||||
| H2O, O2
H2O2, NH3 |
FAD | Monoamino-Oxidase (MAO) | 1.4.3.4 | Ox | MAO A: Brunner-S., Antisozialität | |||||
|
||||||||||
| H2O, NAD+ | Aldehyd-Dehydrogenase (NAD+) | 1.2.1.3 | Ox | |||||||
|
H2O2 |
oder
|
FAD, Mo, Häm | oder
Aldehyd-Oxidase |
1.2.3.1 | Ox | |||||
|
||||||||||
| S-Adenosylmethionin | FAD | Acetylserotonin- O-Methyltransferase | 2.1.1.4 | Tr | ||||||
|
||||||||||





Das biogene Monoamin Serotonin (5-Hydroxytryptamin, 5-HT) spielt im Körper eine wichtige Rolle als Hormon und Neurotransmitter. Die Wirkung entfaltet sich über eine ganze Reihe verschiedener 5-HT-Rezeptor-Subtypen, so dass Serotonin an verschiedenen Orten unterschiedliche Effekte hervorrufen kann. 5-HT-Rezeptoren beeinflussen z.B. die zentrale Informationsverarbeitung, die Thrombozytenaggregation, den Gefäßtonus und die Darmperistaltik. (Näheres dazu siehe in den Büchern der Physiologie.)
Pathologie: Vor allem im Dünndarm und in der Lunge können sog. Karzinoide (neuroendokrine Tumoren) auftreten, die gerne Serotonin bilden. Besonders bei metastasierten Karzinoiden treten dann typische Symptome auf wie Flush, Diarrhö und Herzklappenfibrose. Im Urin können dann häufig erhöhte Konzentrationen an 5-Hydroxyindolessigsäure nachgewiesen werden.
Pharmakologie: Zahllose Medikamente und Toxine wirken am 5-HT-System. Dazu gehören z.B. Buspiron (Anxiolytikum), Triptane (Migränetherapeutika), Sarpogrelat (Thrombozytenaggregationshemmer), Setrone (Antiemetika), Mutterkorn-Alkaloide (Ergotismus) und LSD (Halluzinationen) sowie verschiedene Antidepressiva.
Biosynthese von Melatonin aus Serotonin
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| Acetyl-CoA | Serotonin-N-Acetyltransferase | 2.3.1.87 | Tr | Delayed sleep phase syndrome (DSPS) | ||||||
|
||||||||||
| S-Adenosylmethionin | Acetylserotonin- O-Methyltransferase | 2.1.1.4 | Tr | |||||||
|
||||||||||
| O2, Red. Flavoprotein
H2O, Ox. Flavoprotein |
Häm- Thiolat | Unspezifische Monooxygenase (Cytochrom P450) Leber | 1.14.14.1 | Ox | ||||||
|
||||||||||



Das Hormon Melatonin findet man in allen Lebewesen. Im Menschen wird es vorwiegend in der Epiphyse (Hirnanhangsdrüse) aus Serotonin gebildet. Durch Einwirkung auf verschiedene Regelkreise reguliert es den Tag-Nacht-Rhythmus. Die pulsatile Sekretion von Melatonin erfolgt hauptsächlich nachts, Tageslicht bremst die Ausschüttung.
Abbau von Tryptophan
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| O2
|
Häm | Tryptophan-2,3-Dioxygenase | 1.13.11.11 | Ox | ||||||
| Indolamin-2,3-Dioxygenase | 1.13.11.52 | |||||||||
|
||||||||||
| H2O | Arylformamidase | 3.5.1.9 | Hyd | |||||||
|
||||||||||
| O2, NADPH/H+
H2O, NADP+ |
FAD | Kynurenin-3-Monooxygenase | 1.14.13.9 | Ox | ||||||
|
||||||||||
| H2O | Pyridoxal- phosphat | Kynureninase | 3.7.1.3 | Hyd | Xanthuren- acidurie | |||||
|
||||||||||
| O2
|
Fe | 3-Hydroxyanthranilat-3,4-Dioxygenase | 1.13.11.6 | Ox | ||||||
|
||||||||||
|
|
Aminocarboxymuconat- semialdehyd-Decarboxylase | 4.1.1.45 | Ly | |||||||
|
||||||||||
| H2O, NAD+ | Aminomuconat-semialdehyd-Dehydrogenase | 1.2.1.32 | Ox | |||||||
|
||||||||||
| H2O, NADH/H+ | Aminocarboxymuconat- semialdehyd-Decarboxylase | 1.5.1.- | Ly | |||||||
|
||||||||||





Tryptophan wird zu Alanin und α-Ketoadipat degradiert. Letzteres kann wie beim Lysin-Stoffwechsel beschrieben über dehydrierende Decarboxylierung und β-Oxidation zu Acetyl-CoA abgebaut werden.
Vom 2-Amino-3-carboxymuconat-semialdehyd zweigt die Nicotinsäure-Biosynthese ab. Niacin ist daher als Vitamin semi-essentiell, da es auch aus Tryptophan gebildet werden kann.
Alternative Abbauwege für L-Kynurenin und 3-Hydroxy-L-Kynurenin
BearbeitenFür L-Kynurenin und 3-Hydroxy-L-Kynurenin gibt es noch zwei alternative Abbauwege, die unter pathologischen Bedingungen eine Rolle spielen:
| ⇓ | Subst. | ⇓ | ⇓ | Subst. | ⇓ | Co. | Enzym | EC | EG | Erkr. | |
|---|---|---|---|---|---|---|---|---|---|---|---|

L-Kynurenin |

3-Hydroxy-L-Kynurenin |
||||||||||
| |||||||||||
| α-Ketoglutarat | α-Ketoglutarat | Pyrid- oxal- phosphat | Kynurenin- Amino- transferase | 2.6.1.7 | Tr | ||||||
-2,4-dioxobutanoat.svg)
4-(2-Aminophenyl)- 2,4-dioxobutanoat |
-2,4-dioxobutanoat.svg)
4-(2-Amino-3-hydroxyphenyl)- 2,4-dioxobutanoat |
||||||||||

Kynurenat |

Xanthurenat |
||||||||||
Pathobiochemie: Bei einem Mangel an Pyridoxalphosphat (Vitamin B6) wie auch beim erblichen Kynureninase-Mangel (Xanthurenacidurie) stauen sich L-Kynurenin und 3-Hydroxy-L-Kynurenin an, die dann vermehrt in Kynurenat und Xanthurenat umgewandelt werden und im Urin nachweisbar sind. (Die Kynureninaminotransferase ist zwar auch B6-abhängig, kommt aber im Ggs. zur Kynureninase nicht nur im Zytosol, sondern auch im Mitochondrium vor und soll daher weniger von einem B6-Mangel tangiert werden (?)).
Da Niacin aus Tryptophan gebildet werden kann, treten Niacin-Mangelerkrankungen wie die Pellagra i.d.R. nur bei kombiniertem Protein(Tryptophan)- und Vitaminmangel (Niacin, B6) auf, z.B. bei überwiegender Maisernährung in Entwicklungsländern oder bei Alkoholismus.
Weblinks
Bearbeiten
Histidin-Stoffwechsel
BearbeitenAllgemeines
BearbeitenL-Histidin ist eine nicht-essentielle proteinogene Aminosäure. Die basische Seitenkette enthält einen Imidazol-Ring, ein fünfgliedriger aromatischer Heterozyklus mit zwei Heteroatomen (Stickstoff). Wie alle proteinogenen Aminosäuren dient Histidin als Rohstoff zur Proteinbiosynthese. In Proteinen kann Histidin posttranslational zu 3-Methyl-Histidin methyliert werden. Viele Enzyme enthalten einen Histidyl-Rest im aktiven Zentrum. Im Hämoglobin ist ein Histidyl-Rest an der Komplexierung des Eisenatoms beteiligt.
Daneben kann die aromatische Aminosäure zum biogenen Amin Histamin decarboxyliert werden, das insbesondere von Gewebsmastzellen und basophilen Granulozyten synthetisiert und z.B. im Rahmen von IgE-vermittelten Immunreaktionen (Allergien, Parasitenabwehr) freigesetzt wird. Histamin wirkt bronchospasmogen und fördert die Stickstoffmonoxid-Freisetzung aus Endothelzellen. Im Magen stimuliert es die Säureproduktion. Im ZNS fungiert es als Neurotransmitter (Schlaf-Wach-Rhythmus, Brechreiz).
Wegen des pKs-Wertes der Seitenkette von ca. 6 liegt diese beim physiologischen pH von 7,4 protoniert und deprotoniert vor. So kann Histidin (bzw. das Protein) als Säure-Basen-Puffer wirken und bei enzymatischen Reaktionen Protonen abgeben und aufnehmen.
Im Hämoglobin besetzt ein Histidin-Rest die 5. Koordinationsstelle des Häm-Eisens.
Abbau von Histidin
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||||||
|
|
Histidase (Histidin-Ammoniak-Lyase) | 4.3.1.3 | Ly | Histidinämie | |||||||||||
|
|||||||||||||||
| H2O
|
NAD | Urocanase (Urocanat-Hydratase) | 4.2.1.49 | Ly | Urocanase-Def. | ||||||||||
|
|||||||||||||||
| H2O
H+ |
Imidazolonpropionase (Imidazolonpropionat-Hydrolase) | 3.5.2.7 | Hyd | ||||||||||||
|
|||||||||||||||
| THF | THF | Pyridoxal- phosphat | Glutamat-Formimidoyltransferase | 2.1.2.5 | Tr | FIGLU-urie | |||||||||
|
|||||||||||||||





Pathobiochemie: Bei Folsäuremangel oder Leberschaden ist die Abbaukapazität für Histidin vermindert. Bei hoher oraler Zufuhr der Aminosäure (FIGLU-Test, Histidinbelastungstest) wird dann vermehrt FIGLU im Urin ausgeschieden. Quelle: Roche Lexikon Medizin: FIGLU-Test
Biosynthese von Histamin und Abbau zu Imidazol-4-acetat
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||||||
|
|
Pyridoxalphosphat od. Pyruvat | Histidin-Decarboxylase | 4.1.1.22 | Ly | |||||||||||
| Pyridoxalphosphat | DOPA-Decarboxylase | 4.1.1.28 | AADC-Def. | ||||||||||||
|
|||||||||||||||
| H2O, O2
NH3, H2O2 |
Cu, TPQ | Diamino-Oxidase (DAO) | 1.4.3.6 | Ox | |||||||||||
|
|||||||||||||||
| H2O, NAD+ | Aldehyd-Dehydrogenase (NAD+) | 1.2.1.3 | Ox | ||||||||||||
|
|||||||||||||||
Auch hier taucht wieder eine Reaktionsfolge auf (Modifikation der Seitenkette durch Decarboxylierung, oxidative Deaminierung und Oxidation), die Sie schon vom Stoffwechsel der anderen aromatischen Aminosäuren her kennen.
Alternativer Abbau des Histamins zu Methylimidazolacetat
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| S-Adenosylmethionin | Histamin-N-Methyltransferase | 2.1.1.8 | Tr | |||||||
|
||||||||||
| H2O, O2
NH3, H2O2 |
FAD | Monoamino-Oxidase (MAO) | 1.4.3.4 | Ox | MAO A: Brunner-S., Antisozialität, Autismus | |||||
|
||||||||||
| H2O, NAD(P)+ | Aldehyd-Dehydrogenase (ALDH) (NAD(P)+) | 1.2.1.5 | Ox | |||||||
|
||||||||||



Weblinks
Bearbeiten
Valin-, Leucin- und Isoleucin-Stoffwechsel
BearbeitenAllgemeines
BearbeitenDie essentiellen, proteinogenen Aminosäuren Leucin, Valin und Isoleucin besitzen eine aliphatische, verzweigte, lipophile Seitenkette.
Abbau der verzweigtkettigen Aminosäuren
Bearbeiten| ⇓ | Subst. | (⇑) | ⇓ | Subst. | (⇑) | ⇓ | Subst. | (⇑) | Co. | Enzym | EC/EG | Erkr. | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|||||||||||||||||||
| α-KG | α-KG | α-KG | Pyrid- oxal- Phos. | Verzweigtkettige- Aminosäuren- Transaminase | 2.6.1.42 Tr | ||||||||||||||||
|
|
|
|||||||||||||||||||
| CoA-SH, NAD+
CO2, NADH/H+ |
CoA-SH, NAD+
CO2, NADH/H+ |
CoA-SH, NAD+
CO2, NADH/H+ |
Thiamin- P2 | Verzweigtkettige- α-Ketosäuren- Dehydrogenase- Komplex | 1.2.4.4 Ox | MSUD I | |||||||||||||||
| 2.3.1.168 Tr | MSUD II | ||||||||||||||||||||
| FAD | 1.8.1.4 Ox | MSUD III | |||||||||||||||||||
|
|
|
|||||||||||||||||||
| A
AH2 |
|
A
AH2 |
A
AH2 |
|
A
AH2 |
A
AH2 |
|
A
AH2 |
FAD | 1) Isovaleryl-CoA- Dehydrogenase
2) Butyryl-CoA- Dehydrogenase 3) Acyl-CoA- Dehydrogenase |
1.3.8.4 Ox
|
| |||||||||
|
|
|
|||||||||||||||||||
| ATP, HCO3-
ADP, Pi |
Biotin | Methyl-crotonoyl- CoA-Carboxylase | 6.4.1.4 Lig | MCC1-Def., MCC2-Def. | |||||||||||||||||
|
|||||||||||||||||||||
| H2O
|
1.
|
H2O
|
H2O
|
2.
|
|
H2O
|
2.
|
|
1) Methyl- glutaconyl-CoA- Hydratase
2) Enoyl-CoA- Hydratase |
4.2.1.18 Ly
4.2.1.17 Ly |
1) MGA1
| ||||||||||
|
|
|
|||||||||||||||||||
| H2O | 3-Hydroxy- isobutyryl-CoA- Hydrolase | 3.1.2.4 Hyd | |||||||||||||||||||
|
|||||||||||||||||||||
| NAD+ | 1.,2.
|
NAD+ | 2.,3.
|
NAD+ | 1) 3-Hydroxy- isobutyrat- Dehydrogenase
2) 3-Hydroxy- acyl-CoA- Dehydrogenase 3) 3-Hydroxy- 2-Methylbutyryl- CoA- Dehydrogenase |
1.1.1.31 Ox
|
| ||||||||||||||
|
|
||||||||||||||||||||
| CoA-SH, NAD+
CO2, NADH/H+ |
1.
|
CoA-SH
|
2., 3.
|
1) Methyl- malonat- semialdehyd- Dehydrogenase
2) Acetyl-CoA-C-Acetyltransferase 3) Acetyl-CoA- C-acyltransferase |
1.2.1.27 Ox
2.3.1.9 Tr
2.3.1.16 Tr |
1) MMSDHD
| |||||||||||||||
|
|
||||||||||||||||||||












-3-Hydroxyisobutyryl-CoA.svg)
-3-Hydroxy-2-methylbutyryl-CoA.svg)
-3-Hydroxyisobutyrat.svg)
-Methylmalonatsemialdehyd.svg)


Der Abbau der drei essentiellen verzweigtkettigen Aminosäuren ist sehr ähnlich, zu Anfang steht bei allen eine Transaminierung gefolgt von einer Dehydrierenden Decarboxylierung. Danach folgt die Dehydrierung der zweiten C-C-Bindung.
Der weitere Abbau gestaltet sich wie folgt:
1) L-Leucin-Abbauweg: Methylcrotonoyl-CoA (3-Methylbut-2-enoyl-CoA) wird unter ATP-Verbrauch carboxyliert und dann hydratisiert. Das Endprodukt β-Hydroxy-β-methylglutaryl-CoA (HMG-CoA) kann in die Ketonkörperbiosynthese (und darüber zu Acetyl-CoA abgebaut) oder in die Cholesterinbiosynthese eingeschleust werden.
2) L-Valin-Abbauweg: Methylpropenoyl-CoA wird hydratisiert, CoA-SH wird abgespalten, dann wird es zu Methylmalonatsemialdehyd dehydriert (= oxidiert). Dieses kann über Propionyl-CoA zu Succinyl-CoA abgebaut und in den Citratzyklus eingeschleust werden.
3) L-Isoleucin-Abbauweg: Methylbutenoyl-CoA wird hydratisiert, dehydriert und unter Verwendung eines Coenzym A in Acetyl-CoA und Propionyl-CoA gespalten.
Literatur
Bearbeiten- Harris RA, Joshi M, Jeoung NH, Obayashi M. “Overview of the molecular and biochemical basis of branched-chain amino acid catabolism”. J. Nutr., 135:1527S–30S, June 2005. PMID 15930464.
Weblinks
Bearbeiten
Glutathion-Stoffwechsel
BearbeitenAllgemeines
BearbeitenGlutathion ist ein Tripeptid aus Glutamat, Cystein und Glycin. Es wirkt mit seiner SH-Gruppe vor allem in Erythrozyten als Antioxidans. Für seine Regeneration ist die ständige Zufuhr von NADPH + H+ aus dem Pentosephosphatweg notwendig.
Biosynthese und Abbau von Glutathion (GSH) - der γ-Glutamyl-Zyklus
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| L-Glutamat, ATP
ADP, Pi |
Glutamat--Cystein-Ligase (γ-Glutamyl- cystein-Synthetase) | 6.3.2.2 | Lig | Glutamat--Cystein-Ligase-Defizienz | ||||||
|
||||||||||
| Glycin, ATP
ADP, Pi |
Glutathion-Synthase | 6.3.2.3 | Lig | Hämolyt. Anämie, 5-Ketoprolinurie | ||||||
|
||||||||||
| H2O | γ-Glutamyltransferase (γ-GT) | 2.3.2.2 | Tr | Glutathionurie | ||||||
|
|
oder |
|||||||||
|
||||||||||
| H2O | Zn | Membran-Alanyl-Aminopeptidase | 3.4.11.2 | Hyd | ||||||
|
||||||||||



Glutathion (γ-L-Glutamyl-L-cysteinylglycin, GSH) ist ein Tripeptid aus L-Glutamat, Glycin und L-Cystein. Die Bindung zwischen L-Cystein und L-Glutamat ist dabei etwas ungewöhnlich, da die Peptidbindung hier über die γ-Carboxylgruppe von Glutamat gebildet wird. Dies schützt die Bindung vor der Spaltung durch intrazelluläre Peptidasen.
Die Gamma-Glutamyltransferase (γ-GT) kann den γ-Glutamyl-Rest auch vom Glutathion auf andere Aminosäuren übertragen.
Die Synthese des Tripeptids benötigt keine Ribosomen.
Auf einem Nebenweg entsteht 5-Ketoprolin
BearbeitenAbbau von γ-Glutamylcystein zu Glutamat und Cystein
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| γ-Glutamylcyclotransferase | 2.3.2.4 | Tr | ||||||||
|
||||||||||
| 2 H2O, ATP
ADP, Pi |
5-Ketoprolinase (Pyroglutamase) | 3.5.2.9 | Hyd | 5-Ketoprolinase-Defizienz | ||||||
|
||||||||||
γ-Glutamylcystein wird über zwei enzymatische Schritte unter ATP-Verbrauch zu Cystein und Glutamat gespalten. Zwischenprodukt ist 5-Ketoprolin. Dieser Weg wird insbesondere bei der schweren Glutathion-Synthase-Defizienz (5-Ketoprolinurie) aktiviert. Ansonsten fließt γ-Glutamylcystein überwiegend in die GSH-Biosynthese, da die Glutathion-Synthase für γ-Glutamylcystein einer höheren Affinität aufweist als die γ-Glutamylcyclotransferase.
Abbau von γ-Glutamyl-Aminosäuren zu Glutamat und Aminosäure
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
|
|
γ-Glutamylcyclotransferase | 2.3.2.4 | Tr | |||||||
|
||||||||||
| 2 H2O, ATP
ADP, Pi |
5-Ketoprolinase (Pyroglutamase) | 3.5.2.9 | Hyd | 5-Ketoprolinase-Defizienz | ||||||
|
||||||||||
Die γ-Glutamylcyclotransferase spaltet analog zu oben auch γ-Glutamylreste von anderen Aminosäuren ab, die zuvor im γ-Glutamylzyklus auf die Aminosäuren übertragen wurden (s.o.). Auch dabei entsteht 5-Ketoprolin.
Biologische Funktionen
BearbeitenGlutathion schützt mit seiner reduzierten Sulfhydrylgruppe (SH-Gruppe von Cystein) Hämoglobin in Erythrozyten u.a. Proteine (z.B. in der Leber) vor der Oxidation durch freie Radikale (Hydroxyl-Radikal, Lipidperoxyl-Radikal, Peroxynitrit, H2O2), in dem es sich selbst für Oxidationsvorgänge zur Verfügung stellt, d.h. Elektronen resp. Wasserstoff abgibt. Dadurch kann es z.B. das stark oxidativ wirkende Wasserstoffperoxid (H2O2), das bei verschiedenen Reaktionen frei wird, zu Wasser entgiften. Diese Vorgänge können enzymatisch katalysiert werden oder spontan ablaufen.
Regeneriert wird die oxidierte Form des Glutathions hauptsächlich von der Glutathionreduktase. Die dafür benötigten Elektronen bezieht das Enzym aus NADPH/H+, welches wiederum im 1. Teil des Pentosephosphatwegs aus NADP+ regeneriert wird.
NADP+ kann ebenfalls von der Isocitrat-Dehydrogenase im Citratzyklus zu NADPH/H+ reduziert werden. Diese Möglichkeit haben Erythrozyten wegen der fehlenden Mitochondrien jedoch nicht.
Weitere Aufgaben von Glutathion:
- Glutathion geht in die Biosynthese der Leukotriene C4, D4 und E4 ein.
- Der γ-Glutamyl-Zyklus resp. die γ-Glutamylierung von Aminosäuren dient zum Aminosäuren-Transport in die Zelle.
- Glutathion ist an der Biotransformation Phase II beteiligt.
- Glutathion dient als „Cystein-Transporter“ im Körper.
- Die Glutathionylierung modifiziert die Funktion von verschiedenen Proteinen, z.B. Thioredoxin, Ubiquitin-konjugierendes Enzym und Cytochrom-c-Oxidase.
- Bindung von Stickoxid (NO).
Arbeitsweise von Glutathion als Reduktionsmittel
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| Protein-disulfid
Protein-dithiol |
Protein-disulfid-Reduktase (Glutathion) | 1.8.4.2 | Ox | |||||||
|
2 H2O |
oder
|
Selen | oder
Glutathion-Peroxidase |
1.11.1.9 | Ox | Glutathion-Peroxidase-Def. | ||||
|
||||||||||
| NADPH/H+ | FAD | Glutathion-disulfid-Reduktase | 1.8.1.7 | Ox | Glutathion-disulfid-Reduktase-Def. | |||||
|
||||||||||

Regulation
BearbeitenDie GSH-Produktion und der GSH-Verbrauch resp. die Balance zwischen GSH und GSSG untersteht zahlreichen Einflüssen.
- Die Expression/Aktivität der γ-Glutamylcystein-Synthetase (GCS), des geschwindigkeitsbestimmenden Enzyms, wird durch zahlreiche Faktoren erhöht:
- Oxidativer Stress (via NFκB), nitrosativer Stress
- Inflammation (via NFκB), Krebs, Chemotherapie, ionisierende Strahlung
- Inhibition der GCS-Aktivität
- GSH-Depletion (via NFκB), GSH-Konjugation
- Prostaglandin A2, Schwermetalle, Antioxidantien, Insulin
- Erhöhte Zufuhr von Cystein, Cystin, N-Acetyl-Cystein, Methionin.
- Die Expression/Aktivität der GCS wird vermindert durch:
- Proteinmangel (Glutamat, Cystein, Glycin!)
- Hyperglykämie
- GCS-Phosphorylierung
- Hormone/Zytokine:
- Dexamethason
- Erythropoietin
- TGFß (tumor growth factor ß)
- NO-Anstieg: S-Nitrosation von GCS durch NO-Donatoren. (Link zur Stickoxid-Synthase).
Transport
BearbeitenGSH bzw. γ-Glutamylcystein wird vor allem in der Leber transportiert und dann über den Blutweg an die extrahepatischen Organe (v.a. Niere, Lunge, Darm) geliefert. Wegen des hohen GSH-Gradienten wird GSH schlecht in die Zelle aufgenommen. Daher wird es dort zerlegt und nach Aufnahme in die Zelle neu synthetisiert. γ-Glutamylcystein kann auch direkt aufgenommen werden.
Pathobiochemie
Bearbeiten |
 |
 |
Die Glucose-6-phosphat-Dehydrogenase ist ein Enzym des HMP-Wegs. Ein G6PDH-Mangel führt zum NADPH-Mangel im Erythrozyt, dadurch zur mangelhaften Re-Reduktion des Glutathions und als Folge zu hämolytischen Krisen. Zu ähnlichen Symptomen führen auch andere seltenere Defekte der Glutathion-Stoffwechsel-Enzyme, was die Bedeutung dieses Reduktionssystems für den Erythrozyten unterstreicht.
Die Glutathion-Peroxidase enthält die in Proteinen selten anzutreffende Aminosäure Selenocystein, die im Genom durch ein Stop-Codon (UGA) kodiert wird. Ein Selenmangel, wie er endemisch in einigen Regionen Chinas vorkommt führt zur Keshan-Krankheit, einer schweren Kardiomyopathie. Die Beteiligung des humanen Coxsackievirus B3 an der Pathogenese wird diskutiert.
Klinische Chemie
BearbeitenDie γ-GT (GGT) im Serum ist leber- und gallengangsspezifisch, obwohl das Enzym auch in vielen anderen Geweben vorkommt. Die Bestimmung des γ-GT-Spiegels wird daher in der Leber- und Gallenwegs-Diagnostik genutzt. Das Enzym ist membrangebunden und steigt schon bei geringen zellulären Schäden an. Seine Sensitivität ist damit höher als die der GOT und GPT. Der γ-GT-Spiegel korreliert mit dem Ausmaß der Leberschädigung. Die HWZ beträgt 3-4 Tage.
Weblinks: Laborlexikon - Gamma-GT
Toxikologie
BearbeitenBeim Abbau von Paracetamol in der Leber entsteht das reaktive N-Acetyl-p-benzochinonimin (NAPQI), das normalerweise durch Glutathion unschädlich gemacht wird. Bei einer Paracetamolvergiftung erschöpft sich die Entgiftungskapazität des Glutathions und NAPQI beginnt die Zellbestandteile zu schädigen. Die Symptome des Leberversagens stellen sich oft erst nach einer symptomarmen Latenzzeit von 12 bis 24 h ein. In den ersten Stunden nach der Einnahme ist die Zufuhr von N-Acetylcystein (ACC, NAC) erfolgsversprechend. NAC besitzt ebenfalls SH-Gruppen und kann die Glutathion-Lücke schließen.
Literatur
BearbeitenPMID 14988435 PMID 12818476 PMID 18158646 PMID 10569625 PMID 10385608 PMID 19166318 PMID 18601945
Weblinks
Bearbeiten- KEGG: Glutathione metabolism - Homo sapiens (human)
- PDB Molecule of the Month - Selenocystein-Synthase
Glycan-Stoffwechsel
BearbeitenEigenschaften und biologische Bedeutung
BearbeitenHomoglycane sind Oligo- und Polysaccharide, die nur aus einem einzigen Monosaccharid-Baustein bestehen. Dazu zählen z.B. Stärke, Glycogen und Cellulose. Alle drei bestehen aus Glucose-Molekülen. Bei Stärke und Glycogen handelt es sich um α(1->4)-glykosidisch verbundene Glucose-Moleküle, die α(1->6)-glykosidisch verzweigt sind. Sie werden in den Kapiteln Glycogenolyse und Stärkeabbau und Glycogensynthese besprochen. Cellulose besteht aus unverzweigten β(1->4)-glykosidisch verbundenen Glucose-Molekülen und ist die wichtigste Gerüstsubstanz von Pflanzen. Sie kann vom Menschen nicht verdaut werden, hat aber als Ballaststoff eine wichtige Bedeutung für die Verdauungsfunktion.
In den folgenden Kapiteln geht es um Heteroglycane, die aus verschiedenen Monosaccharid-Einheiten zusammengesetzt sind. Sie sind meist mit Proteinen oder Lipiden verknüpft. Durch die zahlreichen Kombinationsmöglichkeiten der verschiedenen Monosaccharide entsteht ein großes Spektrum an Heteroglycanen, das in seiner Vielfalt durchaus mit der Vielfalt der Proteine vergleichbar ist.
- Glycoproteine bestehen aus einem „normalen“ Protein, das posttranslational mit Oligosacchariden modifiziert wurde, die z.B. als Erkennungssignal dienen. Glycoproteine sind daher meistens Proteine, die in die Membran eingebaut oder sezerniert werden.
- Proteoglycane/Glycosaminoglycane bestehen aus einem einfach aufgebauten Proteinskelett (core protein) und repetitiven Disaccharideinheiten. Sie bilden einen großen Teil der extrazellulären Matrix. Aufgrund der vielen OH-Gruppen können sie gut Wasser binden und dem Gewebe (z.B. Knorpel) Elastizität verleihen. Zu dieser Gruppe gehört z.B. das Keratansulfat, das Chondroitinsulfat und das Heparansulfat.
- Glycolipide finden sich in Zellmembranen. Dazu gehören z.B. die Ganglioside (Sphingolipide mit Oligosaccharid-Anteil), die Blutgruppen-Antigene und die GPI-Anker.
Wichtige Monosaccharide im allgemeinen und im Glycan-Stoffwechsel
BearbeitenHier sehen sie eine Galerie mit den wichtigsten C5- und C6-Monosacchariden des Intermediärstoffwechsels, etwas geordnet nach Struktur und Syntheseweg. (Zur besseren Vergleichbarkeit jeweils Darstellung der α-Pyranoseform, soweit möglich.) Diejenigen, die häufig in Glycanen vorkommen sind farblich unterlegt. Dabei handelt es sich ausschließlich um Aldopentosen, Aldohexosen und (N-Acetyl)-Amino-Derivate der Aldohexosen. D-Galactosamin, D-Mannosamin, N-Acetyl-D-Mannosamin, D-Ribose und D-Fructose (eine Ketohexose) werden scheinbar nicht oder kaum in Glycane eingebaut, ebensowenig wie die Ketopentosen D-Ribulose und D-Xylulose.
| Ketopentosen | 
|

|
||
|---|---|---|---|---|
| D-Ribulose (Rib) | D-Xylulose | |||
| Aldopentosen | 
|

|
||
| D-Ribose (Rib) | D-Xylose (Xyl) | |||
| Oxidierte/ reduzierte Hexosen | 
|

|

|
|
| L-Iduronat (IdoA) | D-Glucuronat (GlcA) | L-Fucose (Fuc) | ||
| Hexosen (Aldo- und Keto-) | 
|

|

|

|
| D-Galactose (Gal) | D-Glucose (Glc) | D-Mannose (Man) | D-Fructose | |
| Hexosamine | 
|

|

|
|
| D-Galactosamin (GalN) | D-Glucosamin (GlcN) | D-Mannosamin (ManN) | ||
| N-Acetyl-Hexosamine | 
|

|

|
|
| N-Acetyl-D-Galactosamin (GalNAc) | N-Acetyl-D-Glucosamin (GlcNAc) | N-Acetyl-D-Mannosamin (ManNAc) | ||
| N-Acetyl-Hexosamin-Derivate | 
|
|||
| N-Acetylneuraminsäure (NANA, Neu5NAc) |
Weblinks
BearbeitenN-Glycan-Stoffwechsel
BearbeitenDie Gruppe der N-Glycane befindet sich in Proteinen an Asparagin (N hier als Abkürzung für Asparagin) gebunden. N-Glycane werden zuerst an Dolicholdiphosphat synthetisiert und dann im Verlauf auf Asparagin übertragen.
Dolichol ist ein Terpenoid und leitet sich vom Farnesylpyrophosphat (2-trans,6-trans-Farnesyldiphosphat) ab.
Biosynthese von Dolicholphosphat
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| Isopentenyl-PP
PPi |
Dehydrodolichol-diphosphat-Synthase | 2.5.1.- | Tr | |||||||
|
||||||||||
| n Isopentenyl-PP
n PPi |
Dehydrodolichol-diphosphat-Synthase | 2.5.1.- | Tr | |||||||
|
||||||||||
| Red. Akz.
Ox. Akz. |
? | 1.-.-.- | Ox | |||||||
|
||||||||||
| H2O
Pi |
Dolichol-Diphosphatase | 3.6.1.43 | Hyd | |||||||
|
||||||||||



Weblinks
Bearbeiten- KEGG: Terpenoid biosynthesis - Homo sapiens (human)
- KEGG: N-Glycan biosynthesis - Homo sapiens (human)
- KEGG: N-Glycan degradation - Homo sapiens (human)
O-Glycan-Stoffwechsel
BearbeitenO-Glycane werden am Serin- oder Threonin-Rest eines Proteins oder Peptids gebildet.
Weblinks
Bearbeiten
Keratansulfat-Stoffwechsel
BearbeitenDie Startmoleküle für die Biosynthese von Keratansulfat entstammen der N-Glycan-Biosynthese (Keratansulfat I) oder der O-Glycan-Biosynthese (Keratansulfat II).
Der Abbau ist im Kapitel Abbau der Glycosaminoglycane dargestellt.
Struktur von Keratansulfat I
Bearbeiten Fucα1
[4Gal(6S)β1-4GlcNAc(6S)β1]0-40-[3Galβ1-4GlcNAc(6S)β1]10-12-[3Galβ1-4GlcNAcβ1]-3Galβ1-4GlcNAcβ1-2Manα1-6 6
Manβ1-4GlcNAcβ1-4GlcNAc-Asn
NeuNAcα2-3Galβ1-4GlcNAcβ1-2Manα1-3
Struktur von Keratansulfat II
Bearbeiten[4Gal(6S)β1-4GlcNAc(6S)β1]1-7-[3Galβ1-4GlcNAc(6S)β1]1-6-[3Galβ1-4GlcNAcβ1]-3Galβ1-4GlcNAcβ1-6
GlcNAcα1-Ser/Thr
NeuNAcα2-3Galβ1-3
Weblinks
Bearbeiten| Farbcode Monosaccharide: |
| Glc (D-Glucose) |
| Gal (D-Galactose) |
| Xyl (D-Xylose) |
| Man (D-Mannose) |
| Fuc (L-Fucose) |
| GlcA (D-Glucuronat) |
| IdoA (L-Iduronat) |
| GalNAc (N-Acetyl-D-Galactosamin) |
| GlcNAc (N-Acetyl-D-Glucosamin) |
| GlcN (D-Glucosamin) |
| Neu5NAc (N-Acetylneuraminsäure) |
Chondroitinsulfat-Stoffwechsel
BearbeitenBiosynthese des Polysaccharidgerüsts
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
| Serin-Rest (Protein) | |||||||
| UDP-α-D-Xylose
UDP |
Protein- Xylosyltransferase | 2.4.2.26 | Tr | ||||
|
4Xylβ1-Ser-R |
|||||||
| UDP-D-Galactose
UDP |
Mn | Galactosyltransferase I | 2.4.1.133 | Tr | Ehlers-Danlos- S., progeroide Form | ||
|
Galβ1-4Xylβ1-Ser-R |
|||||||
| UDP-D-Galactose
UDP |
Mn | Galactosyltransferase II | 2.4.1.134 | Tr | |||
|
Galβ1-3Galβ1-4Xylβ1-Ser-R |
|||||||
| UDP-D-Glucuronat
UDP |
Mn | Glucuronosyltransferase I | 2.4.1.135 | Tr | |||
| UDP-N-Acetyl-D-Galactosamin
UDP |
N-Acetylgalactosaminyl- transferase I | 2.4.1.174 | Tr | ||||
|
GalNAcβ1-4GlcAβ1-3Galβ1-3Galβ1-4Xylβ1-Ser-R |
|||||||
| UDP-D-Glucuronat
UDP |
Chondroitin- Glucuronyltransferase II | 2.4.1.226 | Tr | ||||
|
GlcAβ1-3GalNAcβ1-4GlcAβ1-3Galβ1-3Galβ1-4Xylβ1-Ser-R |
|||||||
| UDP-N-Acetyl-D-Galactosamin
UDP |
N-Acetylgalactosaminyl- transferase II | 2.4.1.175 | Tr | ||||
|
GalNAcβ1-4GlcAβ1-3GalNAcβ1-4GlcAβ1-3Galβ1-3Galβ1-4Xylβ1-Ser-R |
|||||||
...
|
|||||||
|
GalNAcβ1-[4GlcAβ1-3GalNAcβ1]n-4GlcAβ1-3Galβ1-3Galβ1-4Xylβ1-Ser-R |
|||||||
Chondroitinsulfat besteht aus repetitiven Disaccharid-Einheiten aus D-Glucuronat (GlcA) und N-Acetyl-D-Galactosamin (GalNAc). Diese sind über die Trisaccharid-Sequenz D-Galactose (Gal) - D-Galactose (Gal) - D-Xylulose (Xyl) an einem Serin-Rest eines Peptids oder Proteins befestigt.
Nach der Bildung der ersten 4 Zucker-Einheiten (GlcA-Gal-Gal-Xyl-Ser-R) zweigt die Biosynthese von Heparansulfat ab.
Sulfatierung und Modifikation der Disaccharid-Einheiten
BearbeitenDie Disaccharid-Einheiten werden nun verschiedentlich mit weiteren Schwefelgruppen versehen (Bildung von Chondroitinsulfat A, C, D, E) und D-Glucuronat teilweise zu L-Iduronat epimerisiert (Bildung von Chondroitinsulfat B = Dermatansulfat).
Übersicht über die verschiedenen Chondroitinsulfate und die Syntheseenzyme:
| Name | Formel | Co | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|
| Chondroitinsulfat A | R-[4GlcAβ1-3GalNAc(4S)β1]-R | Chondroitin-4-Sulfotransferase | 2.8.2.5 | Tr | ||
| Chondroitinsulfat B (Dermatansulfat) | R-[4IdoAβ1-3GalNAc(4S)β1]-R | Chondroitin-Glucuronat-5-Epimerase | 5.1.3.19 | Iso | ||
| Chondroitin-4-Sulfotransferase | 2.8.2.5 | Tr | ||||
| Chondroitinsulfat C | R-[4GlcAβ1-3GalNAc(6S)β1]-R | Chondroitin-6-Sulfotransferase | 2.8.2.17 | Tr | Spondyloepiphyseale Dysplasie, Typ Omani | |
| Chondroitinsulfat D | R-[4GlcA(2S)β1-3GalNAc(6S)β1]-R | Uronyl-2-Sulfotransferase | 2.8.2.- | Tr | ||
| Chondroitin-6-Sulfotransferase | 2.8.2.17 | Tr | Spondyloepiphyseale Dysplasie, Typ Omani | |||
| Chondroitinsulfat E | R-[4GlcAβ1-3GalNAc(4S,6S)β1]-R | Chondroitin-4-Sulfotransferase | 2.8.2.5 | Tr | ||
| N-Acetylgalactosamin-4-sulfat- 6-O-Sulfotransferase | 2.8.2.33 | Tr | ||||
| R-[4IdoA(2S)β1-3GalNAc(4S)β1]-R | Chondroitin-Glucuronat-5-Epimerase | 5.1.3.19 | Iso | |||
| Uronyl-2-Sulfotransferase | 2.8.2.- | Tr | ||||
| Chondroitin-4-Sulfotransferase | 2.8.2.5 | Tr |
Abbau
BearbeitenSiehe im Kapitel Abbau der Glycosaminoglycane.
| Farbcode Monosaccharide: |
| Glc (D-Glucose) |
| Gal (D-Galactose) |
| Xyl (D-Xylose) |
| Man (D-Mannose) |
| Fuc (L-Fucose) |
| GlcA (D-Glucuronat) |
| IdoA (L-Iduronat) |
| GalNAc (N-Acetyl-D-Galactosamin) |
| GlcNAc (N-Acetyl-D-Glucosamin) |
| GlcN (D-Glucosamin) |
| Neu5NAc (N-Acetylneuraminsäure) |
Weblinks
BearbeitenHeparansulfat-Stoffwechsel
BearbeitenBiosynthese des Polysaccharidgerüsts
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
| UDP-N-Acetyl-D-Glucosamin
UDP |
α-N-Acetylglucosaminyl- transferase I | 2.4.1.223 | Tr | ||||
|
GlcNAcα1-4GlcAβ1-3Galβ1-3Galβ1-4Xylβ1-Ser-R |
|||||||
| UDP-D-Glucuronat
UDP |
Heparan- Glucuronyltransferase II | 2.4.1.225 | Tr | ||||
|
GlcAβ1-4GlcNAcα1-4GlcAβ1-3Galβ1-3Galβ1-4Xylβ1-Ser-R |
|||||||
| UDP-N-Acetyl-D-Glucosamin
UDP |
α-N-Acetylglucosaminyl- transferase II | 2.4.1.224 | Tr | ||||
|
GlcNAcα1-4GlcAβ1-4GlcNAcα1-4GlcAβ1-3Galβ1-3Galβ1-4Xylβ1-Ser-R |
|||||||
...
|
|||||||
|
GlcNAcα1-[4GlcAβ1-4GlcNAcα1]n-4GlcAβ1-3Galβ1-3Galβ1-4Xylβ1-Ser-R |
|||||||
| m H2O, m PAPS | N-Deacetylase | 3.1.1.- | Hyd | ||||
| N-Sulfotransferase | 2.8.2.- 2.8.2.8 |
Tr | |||||
|
GlcNAcα1-[4GlcAβ1-4GlcN(2S)α1]n-4GlcAβ1-3Galβ1-3Galβ1-4Xylβ1-Ser-R |
|||||||
Die Struktur der Kohlenhydratkette ähnelt der von Chondroitinsulfat, wobei N-Acetyl-D-Galactosamin (GalNAc) durch N-Acetyl-D-Glucosamin (GlcNAc) ersetzt ist und die entsprechende Bindung zum D-Glucuronat (GlcA) nun α(1->4) ist statt β(1->4).
Sulfatierung und Modifikation der Disaccharid-Einheiten
BearbeitenAnders als beim Chondroitinsulfat wird das N-Acetyl-D-Glucosamin nun vielfach zum D-Glucosamin deacetyliert und die Aminogruppe mit Hilfe von 3'-Phosphoadenylylsulfat (PAPS) sulfatiert, so dass N-Sulfoglucosamin entsteht (s.o.).
Wiederum wie beim Chondroitinsulfat werden die Disaccharid-Einheiten dann verschiedentlich mit weiteren Schwefelgruppen versehen und D-Glucuronat teilweise zu L-Iduronat epimerisiert.
Übersicht über verschiedene Modifikationsmöglichkeiten und die Syntheseenzyme:
| Formel | Co | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|
| R-[4IdoAβ1-4GlcN(2S,6S)α1]-R | NAD | Heparosan-N-sulfat-glucuronat-5-Epimerase | 5.1.3.17 | Iso | |
| Heparansulfat-6-O-Sulfotransferase 1 oder 3 (HS6ST1, HS6ST3) | 2.8.2.- | Tr | |||
| R-[4IdoA(2S)β1-4GlcN(2S,6S)α1]-R | NAD | Heparosan-N-sulfat-glucuronat-5-Epimerase | 5.1.3.17 | Iso | |
| Heparansulfat-6-O-Sulfotransferase 1 oder 3 (HS6ST1, HS6ST3) | 2.8.2.- | Tr | |||
| Heparansulfat-2-O-Sulfotransferase (HS2ST1) | 2.8.2.- | Tr | |||
| R-[4IdoA(2S)β1-4GlcN(2S,3S)α1]-R | NAD | Heparosan-N-sulfat-glucuronat-5-Epimerase | 5.1.3.17 | Iso | |
| Heparansulfat(-Glucosamin)-3-O-Sulfotransferase 2 (HS3ST2) | 2.8.2.29 | Tr | |||
| Heparansulfat-2-O-Sulfotransferase 1 (HS2ST1) | 2.8.2.- | Tr | |||
| R-[4IdoA(2S)β1-4GlcN(3S)α1]-R | NAD | Heparosan-N-sulfat-glucuronat-5-Epimerase | 5.1.3.17 | Iso | |
| Heparansulfat(-Glucosamin)-3-O-Sulfotransferase 3 (HS3ST3) | 2.8.2.30 | Tr | |||
| Heparansulfat-2-O-Sulfotransferase 1 (HS2ST1) | 2.8.2.- | Tr | |||
| R-[4GlcA(2S)β1-4GlcN(2S)α1]-R | Heparansulfat-2-O-Sulfotransferase 1 (HS2ST1) | 2.8.2.- | Tr | ||
| R-[4GlcAβ1-4GlcN(2S,3S,6S)α1]-R | Heparansulfat(-Glucosamin)-3-O-Sulfotransferase 1 (HS3ST1) | 2.8.2.23 | Tr | ||
| Heparansulfat-6-O-Sulfotransferase 2 (HS6ST2) | 2.8.2.- | Tr | |||
| Heparansulfat-6-O-Sulfotransferase 3 (HS6ST3) | 2.8.2.- | Tr | |||
| R-[4GlcA(2S)β1-4GlcN(2S)α1]-R | Heparansulfat-2-O-Sulfotransferase 1 (HS2ST1) | 2.8.2.- | Tr | ||
| R-[4GlcAβ1-4GlcN(2S,6S)α1]-R | Heparansulfat-6-O-Sulfotransferase 2 (HS6ST2) | 2.8.2.- | Tr | ||
| Heparansulfat-6-O-Sulfotransferase 3 (HS6ST3) | 2.8.2.- | Tr | |||
| R-[4GlcA(2S)β1-4GlcN(2S,3S)α1]-R | Heparansulfat(-Glucosamin)-3-O-Sulfotransferase 2 (HS3ST2) | 2.8.2.29 | Tr | ||
| Heparansulfat-2-O-Sulfotransferase 1 (HS2ST1) | 2.8.2.- | Tr |
| Farbcode Monosaccharide: |
| Glc (D-Glucose) |
| Gal (D-Galactose) |
| Xyl (D-Xylose) |
| Man (D-Mannose) |
| Fuc (L-Fucose) |
| GlcA (D-Glucuronat) |
| IdoA (L-Iduronat) |
| GalNAc (N-Acetyl-D-Galactosamin) |
| GlcNAc (N-Acetyl-D-Glucosamin) |
| GlcN (D-Glucosamin) |
| Neu5NAc (N-Acetylneuraminsäure) |
Abbau
BearbeitenSiehe im Kapitel Abbau der Glycosaminoglycane.
Weblinks
BearbeitenAbbau der Glycosaminoglycane
BearbeitenAbbau von Dermatansulfat (Chondroitinsulfat B)
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
|
R-[4IdoA(2S)β1-3GalNAc(4S)β1-4GlcAβ1-3GalNAc(4S)β1]n-R Dermatansulfat (Chondroitinsulfat B) |
|||||||
| H2O
Dermatansulfat |
3.2.-.- | Hyd | |||||
|
IdoA(2S)β1-3GalNAc(4S)β1-4GlcAβ1-3GalNAc(4S) |
|||||||
| H2O | Iduronat-2-Sulfatase | 3.1.6.13 | Hyd | Mucopolysaccharidose II (Hunter) | |||
|
IdoAβ1-3GalNAc(4S)β1-4GlcAβ1-3GalNAc(4S) |
|||||||
| H2O
L-Iduronat |
L-Iduronidase | 3.2.1.76 | Hyd | Mucopolysaccharidose IH (Hurler), IS (Scheie) und IH/S (Hurler/Scheie) | |||
|
GalNAc(4S)β1-4GlcAβ1-3GalNAc(4S) |
|||||||
| H2O | Arylsulfatase B | 3.1.6.12 | Hyd | Mucopolysaccharidose VI (Maroteaux-Lamy) | |||
|
GalNAcβ1-4GlcAβ1-3GalNAc(4S) |
|||||||
| H2O | Hyalurono- glucosaminidase | 3.2.1.35 | Hyd | Mucopolysaccharidose IX | |||
|
GlcAβ1-3GalNAc(4S) |
|||||||
N-Acetylgalactosamin-4-sulfat kann auch direkt abgespalten werden:
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
|
GalNAc(4S)β1-4GlcAβ1-3GalNAc(4S) |
|||||||
| H2O
N-Acetylgalactosamin-4-sulfat |
Hyalurono- glucosaminidase | 3.2.1.35 | Hyd | Mucopolysaccharidose IX | |||
|
GlcAβ1-3GalNAc(4S) |
|||||||
Abbau von Chondroitinsulfat
BearbeitenDer Abbau entspricht den letzten Schritten des Abbaus von Dermatansulfat.
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
|
R-[3GalNAc(S)β1-4GlcAβ1-3GalNAc(S)β1]-R Chondroitinsulfat |
|||||||
| H2O
Chondroitinsulfat |
3.2.-.- | Hyd | |||||
|
GalNAc(S)β1-4GlcAβ1-3GalNAc(S) |
|||||||
| H2O | Arylsulfatase B | 3.1.6.12 | Hyd | Mucopolysaccharidose VI (Maroteaux-Lamy) | |||
|
GalNAcβ1-4GlcAβ1-3GalNAc(S) |
|||||||
| H2O | Hyalurono- glucosaminidase | 3.2.1.35 | Hyd | Mucopolysaccharidose IX | |||
|
GlcAβ1-3GalNAc(S) |
|||||||
N-Acetylgalactosamin-4-sulfat kann auch hier direkt abgespalten werden:
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
|
GalNAc(S)β1-4GlcAβ1-3GalNAc(S) |
|||||||
| H2O
N-Acetylgalactosamin-4-sulfat |
Hyalurono- glucosaminidase | 3.2.1.35 | Hyd | Mucopolysaccharidose IX | |||
|
GlcAβ1-3GalNAc(S) |
|||||||
Abbau von Heparansulfat
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
|
R-[4IdoA(2S)β1-4GlcN(2S)α1-4GlcA(2S)β1-4GlcNAc(6S)α1-4GlcAβ1]-R Heparansulfat |
|||||||
| H2O
Heparansulfat |
HPSE, HPSE2 | 3.2.-.- | Hyd | ||||
|
IdoA(2S)β1-4GlcN(2S)α1-4GlcA(2S)β1-4GlcNAc(6S)α1-4GlcA |
|||||||
| H2O | Iduronat-2-sulfatase | 3.1.6.13 | Hyd | Mucopoly- saccharidose II (Hunter) | |||
|
IdoAβ1-4GlcN(2S)α1-4GlcA(2S)β1-4GlcNAc(6S)α1-4GlcA |
|||||||
| H2O
L-Iduronat |
L-Iduronidase | 3.2.1.76 | Hyd | Mucopolysaccharidose IH (Hurler), IS (Scheie) und IH/S (Hurler/Scheie) | |||
|
GlcN(2S)α1-4GlcA(2S)β1-4GlcNAc(6S)α1-4GlcA |
|||||||
| H2O | N-Sulfoglucosamin- Sulfohydrolase (Sulfamidase) | 3.10.1.1 | Hyd | Mucopoly- saccharidose IIIA (Sanfilippo A) | |||
|
GlcNα1-4GlcA(2S)β1-4GlcNAc(6S)α1-4GlcA |
|||||||
| Acetyl-CoA
CoA-SH |
Heparan- α-Glucosaminid- N-Acetyltransferase | 2.3.1.78 | Tr | Mucopoly- saccharidose IIIC (Sanfilippo C) | |||
|
GlcNAcα1-4GlcA(2S)β1-4GlcNAc(6S)α1-4GlcA |
|||||||
| H2O | α-N-Acetyl- glucosaminidase | 3.2.1.50 | Hyd | Mucopoly- saccharidose IIIB (Sanfilippo B) | |||
|
GlcA(2S)β1-4GlcNAc(6S)α1-4GlcA |
|||||||
| H2O | Glucuronat- 2-Sulfatase | 3.1.6.18 | Hyd | ||||
|
GlcAβ1-4GlcNAc(6S)α1-4GlcA |
|||||||
| H2O | β-Glucuronidase | 3.2.1.31 | Hyd | Mucopoly- saccharidose VII | |||
|
GlcNAc(6S)α1-4GlcA |
|||||||
| H2O | N-Acetylglucosamin- 6-sulfatase | 3.1.6.14 | Hyd | Mucopoly- saccharidose IIID (Sanfilippo D) | |||
|
GlcNAcα1-4GlcA |
|||||||
Abbau von Keratansulfat
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
|
R-[3Gal(6S)β1-4GlcNAc(6S)β1-3Galβ1-4GlcNAc(6S)β1-3Galβ1]-R |
|||||||
| H2O
Keratansulfat |
Keratansulfat-Endo- 1,4-β-galactosidase | 3.2.1.103 | Hyd | ||||
|
Gal(6S)β1-4GlcNAc(6S)β1-3Galβ1-4GlcNAc(6S)β1-3Gal |
|||||||
| H2O | N-Acetylgalactosamin- 6-sulfatase | 3.1.6.4 | Hyd | Mucopoly- saccharidose IVA (Morquio A) | |||
|
Galβ1-4GlcNAc(6S)β1-3Galβ1-4GlcNAc(6S)β1-3Gal |
|||||||
| H2O | β-Galactosidase | 3.2.1.23 | Hyd | GM1-Gangliosidose, Mucopolysaccharidose IVB (Morquio B) | |||
|
GlcNAc(6S)β1-3Galβ1-4GlcNAc(6S)β1-3Gal |
|||||||
| H2O | N-Acetylglucosamin- 6-sulfatase | 3.1.6.14 | Hyd | Mucopoly- saccharidose IIID (Sanfilippo D) | |||
|
GlcNAcβ1-3Galβ1-4GlcNAc(6S)β1-3Gal |
|||||||
| H2O | β-N-Acetyl- hexosaminidase | 3.2.1.52 | Hyd | GM2-Gangliosidose I (Tay-Sachs), II (Sandhoff) | |||
|
Galβ1-4GlcNAc(6S)β1-3Gal |
|||||||
N-Acetyl-D-Glucosamin-6-sulfat kann auch direkt abgespalten werden:
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
|
GlcNAc(6S)β1-3Galβ1-4GlcNAc(6S)β1-3Gal |
|||||||
| H2O
N-Acetyl-D-Glucosamin-6-sulfat |
β-N-Acetylhexosaminidase | 3.2.1.52 | Hyd | GM2-Gangliosidose I (Tay-Sachs), II (Sandhoff) | |||
|
Galβ1-4GlcNAc(6S)β1-3Gal |
|||||||
| Farbcode Monosaccharide: |
| Glc (D-Glucose) |
| Gal (D-Galactose) |
| Xyl (D-Xylose) |
| Man (D-Mannose) |
| Fuc (L-Fucose) |
| GlcA (D-Glucuronat) |
| IdoA (L-Iduronat) |
| GalNAc (N-Acetyl-D-Galactosamin) |
| GlcNAc (N-Acetyl-D-Glucosamin) |
| GlcN (D-Glucosamin) |
| Neu5NAc (N-Acetylneuraminsäure) |
Weblinks
BearbeitenGlycosyl-Phosphatidylinositol-Anker
BearbeitenBiosynthese eines Glycosyl-Phosphatidylinositol-Ankers (GPI-Anker)
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
| UDP-N-Acetyl-D-Glucosamin
UDP |
Phosphatidylinositol- N-Acetylglucosaminyltransferase | 2.4.1.198 | Tr | Paroxysmale nächtliche Hämoglobinurie (PNH) | |||
|
GlcNAcα1-6Ino1-P-Lipid |
|||||||
| H2O | N-Acetylglucosaminyl- phosphatidylinositol-Deacetylase | 3.5.1.89 | Hyd | ||||
|
GlcNα1-6Ino1-P-Lipid |
|||||||
| Palmitoyl-CoA
CoA-SH |
PIG-W | 2.3.-.- | Tr | ||||
|
GlcNα1-6Ino(palmitoyl)1-P-Lipid |
|||||||
| Dolichyl-phosphat-D-Mannose
Dolichyl-phosphat |
PIG-M, PIG-X | 2.4.1.- | Tr | ||||
|
Manα1-4GlcNα1-6Ino(palmitoyl)1-P-Lipid |
|||||||
| Phosphatidylethanolamin
Diacylglycerin |
PIG-N | 2.7.-.- | Tr | ||||
|
EtN-P |
|||||||
| Dolichyl-phosphat-D-Mannose
Dolichyl-phosphat |
PIG-V | 2.4.1.- | Tr | ||||
|
EtN-P |
|||||||
| Dolichyl-phosphat-D-Mannose
Dolichyl-phosphat |
PIG-B | 2.4.1.- | Tr | ||||
|
EtN-P |
|||||||
| Phosphatidylethanolamin
Diacylglycerin |
PIG-F, PIG-O | ? | Tr | ||||
|
EtN-P EtN-P |
|||||||
| Protein
H2O |
GPAA1, PIG-K, PIG-S, PIG-T, PIG-U | ? | ? | ||||
|
H2N-Protein-CO-EtN-P EtN-P |
|||||||
| H2O
Fettsäure |
GPI-Deacylase | ? | ? | ||||
|
H2N-Protein-CO-EtN-P EtN-P |
|||||||
| Farbcode Monosaccharide: |
| Glc (D-Glucose) |
| Gal (D-Galactose) |
| Xyl (D-Xylose) |
| Man (D-Mannose) |
| Fuc (L-Fucose) |
| GlcA (D-Glucuronat) |
| IdoA (L-Iduronat) |
| GalNAc (N-Acetyl-D-Galactosamin) |
| GlcNAc (N-Acetyl-D-Glucosamin) |
| GlcN (D-Glucosamin) |
| Neu5NAc (N-Acetylneuraminsäure) |
GPI-Anker werden in 10 enzymatisch katalysierten Reaktionen im endoplasmatischen Retikulum gebildet und dort an das Carboxyl-Ende von Proteinen geheftet, die die entsprechende Signalsequenz aufweisen.
Biologische Bedeutung
BearbeitenGPI-Anker dienen der Verankerung von Proteinen (z.B. Enzymen wie der Acetylcholinesterase oder Komplementschutzfaktoren) an der Zellmembran, wo sie als sog. „lipid rafts“ organisiert sind. GPI-Anker bestehen aus einem Phosphatidylinositol, wobei das Inositol ein Oligosaccharid trägt, an dem das Protein über ein Ethanolaminphosphat befestigt ist. Das Inositol kann zusätzlich mit einer weiteren Fettsäure (Palmitat) verestert sein.
Pathobiochemie
BearbeitenEine Mutation im Gen der Phosphatidylinositol N-Acetylglucosaminyltransferase verhindert die GPI-Anker-Biosynthese und ist verantwortlich für das Krankheitsbild der Paroxysmalen nächtlichen Hämoglobinurie (PNH).
Literatur
BearbeitenWeblinks
Bearbeiten- KEGG: Glycosylphosphatidylinositol(GPI)-anchor biosynthesis - Homo sapiens (human)
- FIGURE 1 | GPI-anchored-protein precursor and anchor structure. Mayor S, Riezman H. “Sorting GPI-anchored proteins”. Nat. Rev. Mol. Cell Biol., 5:110–20, February 2004. PMID 15040444.
Lacto-Serie
Bearbeiten
Die Blutgruppensysteme ABO und Lewis
BearbeitenZur Laktoserie der Glycosphingolipide gehören u.a. die Blutgruppenmerkmale des AB0-Systems. Man unterscheidet die Merkmale H, A und B, die kodominant vererbt werden. Menschen mit der Blutgruppe 0 tragen das Merkmal H. Bei der Blutgruppe AB findet man sowohl A als auch B auf den Erythrozyten. Da ähnliche Oligosaccharidmuster ubiquitär vorkommen, entwickelt der Organismus IgM-Antikörper gegen die Blutgruppenmerkmale, die er nicht selbst auf seinen Erythrozyten trägt. Gegen H werden keine Antikörper gebildet (daher Blutgruppe "0").
In den Vorstufen der roten Blutzellen werden die AB0-Antigene gebildet. Zuerst wird H synthetisiert und bei der Blutgruppe 0 bleibt es dabei. Träger der Allele A und/oder B, die sich im Besitz der entsprechenden Enzyme befinden, ergänzen das Oligosaccharid dann mit einem N-Acetyl-Galactosamin (A) oder mit einer Galactose (B).
Ein weiteres Blutgruppensystem umfasst die Lewis-Antigene, die sich von den Merkmalen H, A und B einerseits und vom H-Vorläufer ((Neo)Lactosylceramid bzw. (n)Lc4Cer) ableiten, indem zusätzlich Fucose an das N-Acetylglucosamin (GlcNAc) gehängt wird. Die Synthese erfolgt dementsprechend hauptsächlich durch Fucosyltransferasen und zwar außerhalb der Erythropoese. Die Antigene werden nachträglich von außen aufgenommen und in die Zellmembran integriert. Aus dem H-Precursor wird durch Fucosylierung Lewis a, aus dem Antigen H wird Lewis b. Die Lewis-Antigene a und b werden chemisch der Lacto-Serie zugeordnet. Zwei weitere Lewis-Antigene x und y leiten sich analog von der Neo-Lacto-Serie ab. Der wesentliche Unterschied zwischen beiden bzw. zwischen Lacto-Serie und Neo-Lacto-Serie liegt in der Verknüpfung der Galactose (dem 4. Zucker) mit N-Acetylglucosamin (dem 3. Zucker) (Lacto: -Galβ1-3GlcNAc- vs. Neo-Lacto: -Galβ1-4GlcNAc-). Dementsprechend wird die Fucose dann entweder an das freie C4-Atom (Lacto) oder an das freie C3-Atom (Neo-Lacto) des N-Acetylglucosamins addiert. Die Aufteilung in Lacto- und Neo-Lacto-Serie besteht schon bei dem H-Precursor, H, A und B, für die AB0-Blutgruppe ist der Unterschied jedoch irrelevant.
Als Besonderheit kann Lewis a und x weiterhin mit N-Acetylneuraminsäure (Neu5NAc), einer Sialinsäure, versehen sein, die an den freien Galactose-Rest gehängt wird. Lewis x kann repetitive Muster aufweisen.
Medizinische Bedeutung
BearbeitenDas AB0-System ist für die Transfusions- und Transplantationsmedizin sehr bedeutsam. I.d.R werden Blutprodukte und Organe blutgruppengleich übertragen. Ausnahmsweise können Blutprodukte auch entgegen der Blutgruppe übertragen werden, hier sind jedoch nur bestimmte Kombinationen möglich, die auch davon abhängen, ob man zelluläre Bestandteile wie z.B. Erythrozytenkonzentrate (Antigen-haltig) oder Plasma wie z.B. FFP (Antikörper-haltig) überträgt.
Etwa 80 % der Menschen scheiden die Blutgruppenmerkmale des AB0-Systems in ihren Körpersekreten (Speichel, Sperma) aus. Der Ausscheiderstatus korreliert mit dem Lewis-System. Leb-positive sind meist Sekretoren, Lea-positive eher Nicht-Sekretoren, Le-negative entsprechen dem Durchschnitt (80 % Sekretoren). Die Ausscheideeigenschaft (Se) der sog. Sekretoren ist inbesondere für Rechtsmediziner interessant, da die Blutgruppenbestimmung aus Körpersekreten zu Identifikationszwecken genutzt werden kann.
Das Glykoprotein CA 19-9 wird als Tumormarker genutzt, z.B. beim Pankreaskarzinom. Le-negative Menschen (ca. 5 % der Menschen) bilden kein CA 19-9.
Quellen: Medizinische Laboratorien Düsseldorf GbR
Biosynthese der Antigene H und Lewis b (Leb)
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
| UDP-N-Acetyl-D-Glucosamin
UDP |
β-Acetylglucosaminyltransferase | 2.4.1.206 | Tr | ||||
|
GlcNAcβ1-3Galβ1-4Glcβ1-1'Cer |
|||||||
| UDP-D-Galactose
UDP |
β-1,3-Galactosyltransferase 1/2/5 | 2.4.1.- | Tr | ||||
|
Lc4Cer Galβ1-3GlcNAcβ1-3Galβ1-4Glcβ1-1'Cer |
|||||||
| GDP-L-Fucose
GDP |
α-2-L-Fucosyltransferase | 2.4.1.69 | Tr | ||||
|
H Fucα1-2Galβ1-3GlcNAcβ1-3Galβ1-4Glcβ1-1'Cer |
|||||||
| GDP-L-Fucose
GDP |
α-4-L-Fucosyltransferase | 2.4.1.65 | Tr | ||||
|
Fucα1 |
|||||||
Aus Antigen H werden die Antigene A und ALeb gebildet
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
|
H Fucα1-2Galβ1-3GlcNAcβ1-3Galβ1-4Glcβ1-1'Cer |
|||||||
| UDP-N-Acetyl-D-Galactosamin
UDP |
α-N-Acetylgalactosaminyltransferase | 2.4.1.40 | Tr | ||||
|
GalNAcα1 |
|||||||
| GDP-L-Fucose
GDP |
α-4-L-Fucosyltransferase | 2.4.1.65 | Tr | ||||
|
GalNAcα1 Fucα1 |
|||||||
Aus Antigen H werden auch die Antigene B und BLeb synthetisiert
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
|
H Fucα1-2Galβ1-3GlcNAcβ1-3Galβ1-4Glcβ1-1'Cer |
|||||||
| UDP-D-Galactose
UDP |
α-D-Galactosyltransferase | 2.4.1.37 | Tr | ||||
|
Galα1 |
|||||||
| GDP-L-Fucose
GDP |
α-4-L-Fucosyltransferase | 2.4.1.65 | Tr | ||||
|
Galα1 Fucα1 |
|||||||
Aus dem H-Precursor werden die Lewis a-Antigene Lea und Sialyl-Lea gebildet
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
|
Lc4Cer Galβ1-3GlcNAcβ1-3Galβ1-4Glcβ1-1'Cer |
|||||||
| GDP-L-Fucose
GDP |
α-4-L-Fucosyltransferase | 2.4.1.65 | Tr | ||||
|
Fucα1 |
|||||||
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
|
Lc4Cer Galβ1-3GlcNAcβ1-3Galβ1-4Glcβ1-1'Cer |
|||||||
| CMP-N-Acetylneuraminat
CMP |
Sialyltransferase | 2.4.99.6 | Tr | ||||
| β-Galactosid-α-2,3-Sialyltransferase | 2.4.99.4 | Tr | |||||
|
sLc4Cer Neu5Acα2-3Galβ1-3GlcNAcβ1-3Galβ1-4Glcβ1-1'Cer |
|||||||
| GDP-L-Fucose
GDP |
α-4-L-Fucosyltransferase | 2.4.1.65 | Tr | ||||
|
Fucα1 |
|||||||
Übersicht über die Verwandtschaft der AB0- und Lewis-Antigene
Bearbeiten| ABO-Antigene und H-Precursor (Lacto- und Neo-Lacto-Serie) |
Lewis-Antigene (Lacto-Derivate) |
Lewis-Antigene (Neo-Lacto-Derivate) |
|---|---|---|
| Lc4Cer / nLc4Cer | Lea und Sialyl-Lea | Lex (monomer, dimer, trimer) und Sialyl-Lex |
| H | Leb | Ley |
| A | ALeb | / |
| B | BLeb | / |
Vom H-Antigen leiten sich die Lewis-Antigene b und y ab. Vom (Neo)Lactosylceramid stammen Lewis a und x.
Die Blutgruppenmerkmale der Lacto- und Neo-Lacto-Serie im direkten Vergleich
Bearbeiten| ABO-System (Lacto- und Neo-Lacto-Serie) | |
|---|---|
| Lc4Cer / nLc4Cer |
Galβ1-3/4GlcNAcβ1-3Galβ1-4Glcβ1-1'Cer |
| H |
Fucα1-2Galβ1-3/4GlcNAcβ1-3Galβ1-4Glcβ1-1'Cer |
| A |
GalNAcα1 |
| B |
Galα1 |
| Lewis-Antigene der Lacto-Serie | |
| Lea und Sialyl-Lea |
Fucα1 |
| Leb |
Fucα1 |
| ALeb |
GalNAcα1 Fucα1 |
| BLeb |
Galα1 Fucα1 |
| Lewis-Antigene der Neo-Lacto-Serie | |
| Lex |
Fucα1 Fucα1 Fucα1 |
| Dimeres Lex |
Fucα1 Fucα1 |
| Trimeres Lex |
Fucα1 Fucα1 Fucα1 |
| Sialyl-Lex |
Fucα1 |
| Ley |
Fucα1 |
| Farbcode Monosaccharide: |
| Glc (D-Glucose) |
| Gal (D-Galactose) |
| Xyl (D-Xylose) |
| Man (D-Mannose) |
| Fuc (L-Fucose) |
| GlcA (D-Glucuronat) |
| IdoA (L-Iduronat) |
| GalNAc (N-Acetyl-D-Galactosamin) |
| GlcNAc (N-Acetyl-D-Glucosamin) |
| GlcN (D-Glucosamin) |
| Neu5NAc (N-Acetylneuraminsäure) |
Weblinks
Bearbeiten- KEGG: Glycosphingolipid biosynthesis - lactoseries - Homo sapiens (human)
- RCSB PDB: ABO Blood Type Glycosyltransferases
- Laborlexikon - AB0-System
- dbRBC - Lewis Blood Group System
- ABO Blood Group System
Neo-Lactoserie
BearbeitenZu den Unterschieden zwischen den Blutgruppenantigenen der Lacto- und Neo-Lactoserie siehe im Kapitel Lacto-Serie.
Weblinks
Bearbeiten
Globosoide
BearbeitenWeblinks
Bearbeiten
Ganglioside
BearbeitenWeblinks
Bearbeiten
Vitamin-Stoffwechsel
BearbeitenEigenschaften und biologische Bedeutung
BearbeitenVitamine sind organische Substanzen, die der Körper meist in geringer Menge benötigt und die von außen zugeführt werden müssen. Meist dienen sie als Cofaktoren bestimmter Enzyme oder sind anderweitig an chemischen Reaktionen beteiligt. Da sie in der Nahrung meist ausreichend vorhanden sind hat der Organismus aus energetischen Gründen die entsprechenden Vitaminsynthese-Enzyme verloren. Einige Vitamine kann der menschliche Körper zum Teil noch bilden, z.B. Nikotinsäure aus Tryptophan und Vitamin D-Hormon unter Einwirkung von Sonnenlicht aus Cholesterin.
Thiamin-Stoffwechsel
Bearbeiten |
 |
 |
 |
Allgemeines
BearbeitenThiamindiphosphat ist als Cofaktor an der dehydrierenden Decarboxylierung von α-Ketosäuren beteiligt. Weiterhin fungiert es im HMP-Weg als Coenzym der Transketolase.
Biosynthese / Herkunft
BearbeitenDas wasserlösliche schwefelhaltige Vitamin B1 wird von Pflanzen und Bakterien produziert. Es enthält einen Pyrimidin- und einen Thiazol-Ring. Es kann im Menschen als Thiamin, Thiamindiphosphat oder Thiamintriphosphat vorliegen, in Bakterien auch als Thiaminmonophosphat. Die Phosphorylierung von Thiamin erfolgt in Eukaryoten durch die Thiaminpyrophosphokinase.
| ⇓ | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| ATP
AMP |
ATP
AMP |
Thiaminpyrophosphokinase | 2.7.6.2 | Tr | ||||||
|
||||||||||
Biologische Funktionen
Bearbeiten- Thiamindiphosphat (Thiaminpyrophosphat, TPP) ist Coenzym bei Multienzymkomplexen, die die dehydrierende Decarboxylierung (oxidative Decarboxylierung) von α-Ketosäuren katalysieren, dazu gehören:
- Pyruvatdehydrogenase (Dehydrierende Decarboxylierung von Pyruvat zu Acetyl-Coenzym A)
- α-Ketoglutarat-Dehydrogenase (Citratzyklus und Lysin-Abbau)
- Verzweigtkettige-α-Ketosäuren-Dehydrogenase (Valin-, Leucin- und Isoleucin-Abbau).
- Thiamindiphosphat (TPP) ist weiterhin ein Coenzym der Transketolase im Hexosemonophosphatweg
- Im Nervensystem wirkt Thiamin antagonistisch zum Acetylcholin.
Pathobiochemie
BearbeitenEin Thiamin-Mangel kann zu Muskelatrophie, Herzinsuffizienz, Übelkeit, Erbrechen, Leistungsschwäche, Nerven- und Hirnleistungsstörungen führen. Klassisch als Beri-Beri (Ursache: überwiegende Ernährung über polierten Reis in Asien), bei Alkoholikern überwiegend neurologisch als Wernicke-Korsakow-Enzephalopathie. U.U. auch in der Schwangerschaft. Eine weitere Manifestation des Thiamin-Mangels ist die metabolische Azidose durch gesteigerte Lactat-Bildung (Lactat-Azidose).
Weblinks
BearbeitenRiboflavin-Stoffwechsel
BearbeitenAllgemeines
BearbeitenRiboflavin ist als Coenzym der wasserstoffübertragenden Flavoproteine FMN und FAD an Redox-Vorgängen resp. Wasserstoffübertragungen beteiligt.
Biosynthese und Formen der Riboflavine
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| ATP
ADP |
1. |
Pi
H2O |
1) Riboflavin-Kinase | 2.7.1.26 | Tr | |||||
| 2) Saure Phosphatase | 3.1.3.2 | Hyd | ||||||||
|
||||||||||
| ATP
PPi |
1. |
AMP
H2O |
1) FMN-Adenylyltransferase | 2.7.7.2 | Tr | |||||
| 2) Nucleotid-Diphosphatase | 3.6.1.9 | Hyd | ||||||||
|
||||||||||



Riboflavin (Vitamin B2) ist das Coenzym der wasserstoffübertragenden Flavoproteine (Flavinenzyme). Biologisch aktiv ist das Flavinmononucleotid (FMN, Riboflavinphosphat) und das Flavinadenindinucleotid (FAD), das aus Flavin, zwei Phosphatgruppen, Ribose und Adenin besteht. Die Phosphorylierung zur biologisch aktiven Form erfolgt in der Darmmucosa.
Die Biosynthese in Pflanzen und vielen Mikroorganismen erfolgt aus GTP (Purin) und Ribulose-5-phosphat (HMP-Weg).
Biologische Funktionen
BearbeitenFlavinmononucleotid (FMN) ist ein Bestandteil vom Komplex I der Atmungskette.
Flavinadenindinucleotid (FAD) ist Cofaktor von Flavoproteinen, die z.B. folgende Reaktionen katalysieren:
- Oxidative Deaminierung aromatischer proteinogener L-Aminosäuren und ihrer Derivate (Katecholamine, Serotonin, Histamin) durch Monoaminooxidasen (Phenylalanin-, Tyrosin-/Katecholamin-, Tryptophan- und Histidin-Stoffwechsel).
- Als Cofaktor der 3-Isopentenylpyrophosphat-Isomerase und Squalen-Monooxygenase Beteiligung an der Cholesterin-Biosynthese.
- Dehydrierung von Ethan- zu Ethengruppen (C-C <-> C=C + 2H), Die Acyl-CoA-Dehydrogenase katalysiert z.B. den 1. Schritt der β-Oxidation der Fettsäuren.
- Oxidation von Aldehyden zu Säuren, z.B. durch die Xanthinoxidase beim Purin-Abbau.
- Transhydrogenierungen, wie z.B. durch die Dihydrolipoyldehydrogenase, ein Bestandteil der verschiedenen Dehydrogenase-Komplexe:
- Pyruvatdehydrogenase (Dehydrierende Decarboxylierung von Pyruvat zu Acetyl-Coenzym A)
- α-Ketoglutarat-Dehydrogenase (Citratzyklus und Lysin-Abbau)
- Succinat-Dehydrogenase (Citratzyklus bzw. Komplex II der Atmungskette)
- Dehydrogenasen des (Valin-, Leucin- und Isoleucin-Abbaus).
Bei diesen Reaktionen übernimmt eins der vier Stickstoffatome des Flavins ein Hydridanion (H- = H+ + 2 e-), ein anderes ein Proton (H+), d.h. netto werden 2 Wasserstoffatome (2 [H]) übertragen.
Funktionsweise (Aufnahme und Abgabe von Wasserstoff)
Bearbeiten| ⇓ | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| H-, H+
|
H-, H+
|
Ox | ||||||||
|
||||||||||

Pathobiochemie
BearbeitenVitamin B2-Mangelerkrankungen treten selten isoliert auf. Zu den Symptomen gehören Entzündungen der Haut (Mundwinkelrhagaden, seborrhoische Dermatitis, Landkartenzungen-Glossitis) und Hornhaut. Weiterhin sind Wachstumsstörungen, neurologische Störungen und Blutbildungsstörungen möglich.
Weblinks
Bearbeiten
Nicotinat- und Nicotinamid-Stoffwechsel
BearbeitenAllgemeines
BearbeitenNikotinsäure (Niacin, Vitamin B3) ist als Bestandteil von Nicotinamid-adenin-dinucleotid(-phosphat), kurz NAD(P)+ an vielen Oxidoreduktase-Reaktionen in fast allen Stoffwechselwegen in der Zelle beteiligt. Die Ausgangsstoffe für die NAD-Synthese können z.T. aus dem Tryptophan-Stoffwechsel bezogen werden und sind damit semi-essentiell.
Biosynthese aus Tryptophan-Abbauprodukten
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
|
||||||||||
| PRPP
CO2, PPi |
Chinolinat-Phosphoribosyltransferase (carboxylierend) | 2.4.2.19 | Tr | |||||||
|
||||||||||
| ATP
PPi |
1. |
AMP
H2O |
1) Nicotinamid-nucleotid-Adenylyltransferase | 2.7.7.1 | Tr | |||||
| 2) Nucleotid-Diphosphatase | 3.6.1.9 | Hyd | ||||||||
| 2) Deamino-NAD+-Nucleotidohydrolase | 3.6.1.22 | Hyd | ||||||||
|
||||||||||
| L-Glutamin, ATP, H2O
L-Glutamat, AMP, PPi |
NAD+-Synthase (Glutamin-hydrolysierend) | 6.3.5.1 | Lig | |||||||
|
||||||||||
| ATP
AMP |
1. |
Pi
H2O |
1) NAD+-Kinase | 2.7.1.23 | Tr | |||||
| 2) Phosphor-monoester-Hydrolase | 3.1.3.- | Hyd | ||||||||
| oder
|
oder
NAD(P)+-Transhydrogenase (AB-spezifisch) |
1.6.1.2 | Ox | |||||||
|
||||||||||





Der Nikotinsäure-Bestandteil von NAD+ wird zu 2/3 so wie oben dargestellt aus dem Tryptophan-Stoffwechsel bezogen, daher kann es nur bei einem kombinierten Niacin-Tryptophan-Mangel zu Unterversorgungserscheinungen kommen, z.B. bei sehr einseitiger Ernährung auf der Basis von Mais (Entwicklungsländer).
Biosynthese aus Niacin/Nicotinamid
BearbeitenEin zweiter Biosyntheseweg ist der so genannte 'Nicotinamide salvaging pathway', über den beim Abbau anfallendes oder auch mit der Nahrung aufgenommenes Niacin und Nicotinamid verwertet werden können. Man vermutet außerdem die Existenz einer Nicotinamid-Deaminase im menschlichen Stoffwechsel, die ein Gleichgewicht zwischen Niacin und Nicotinamid aufrechterhalten würde. Dies ist aber nicht zwingend notwendig, da beide Stoffe zum Nukleotid und anschließend zum Dinukleotid umgewandelt werden können.
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| PRPP
PPi |
PRPP
PPi |
Nicotinat-Phosphoribosyltransferase | 2.4.2.11 | Tr | ||||||
|
||||||||||

Die Biosynthese aus Nicotinamid läuft parallel zur Biosynthese aus Chinolinat und Nicotinat; der Unterschied besteht darin, dass die Amidogruppe nicht mehr hinzugefügt werden muss.
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| PRPP
PPi |
PRPP
PPi |
Nicotinamid-Phosphoribosyltransferase | 2.4.2.12 | Tr | ||||||
|
||||||||||
| ATP
PPi |
1. |
AMP
H2O |
1) Nicotinamid-nucleotid-Adenylyltransferase | 2.7.7.1 | Tr | |||||
| 2) Nucleotid-Diphosphatase | 3.6.1.9 | Hyd | ||||||||
| 2) NAD+-Diphosphatase | 3.6.1.22 | Hyd | ||||||||
|
||||||||||

Zum Abbau siehe: KEGG: Nicotinate and nicotinamide metabolism - Homo sapiens (human).
Biologische Funktionen
Bearbeiten |
 |
- Nikotinsäure ist Bestandteil der wasserstoffübertragenden Coenzyme Nicotinamid-adenin-dinucleotid (NAD+) und Nicotinamid-adenin-dinucleotid-phosphat (NADP+), die für zahlreiche Oxidoreduktasereaktionen benötigt werden. Die energiereiche Elektronen stammen überwiegend aus katabolen Prozessen wie z.B. der Glycolyse, dem Citratzyklus oder der β-Oxidation. Die Reduktionsäquivalente enthalten ebenso wie ATP chemische Energie, die für Reduktionen in Biosynthesen oder zur ATP-Regenerierung verwendet werden kann. NADH/H+ liefert seine Elektronen bevorzugt in die Atmungskette. NADPH/H+ stellt die Reduktionsäquivalente z.B. der Fettsäuren- und der Cholesterin-Biosynthese zu Verfügung. NADPH/H+ wird hauptsächlich im Hexosemonophosphatweg generiert, kann aber auch unter ATP-Verbrauch im Citrat-Malat-Pyruvat-Zyklus aus NADH/H+ gebildet werden.
NAD(P)+ kann mit seiner Nicotinsäureamid-Struktur formal zwei Wasserstoff-Atome (H) aufnehmen. Wasserstoff-Atome bestehen jeweils aus einem Proton (H+) und einem Elektron (e–). Nimmt NAD(P)+ 2 H-Atome (= 2 H+ + 2 e– = 1 Hydridion H– + H+) und damit 2 e– auf, dann entspricht dies einer Reduktion des NAD(P)+ zum NAD(P)H + H+ (und einer Oxidation und Dehydrierung des H-abgebenden Moleküls). Umgekehrt wird NAD(P)H + H+ oxidiert, wenn es seinen Wasserstoff an andere Moleküle abgibt, die dabei dann reduziert werden.
Das NAD(P)+-Molekül selbst bindet nur ein Hydridion H– (= 1 H+ + 2 e– bzw. 1 H + 1 e–), das überzählige Proton (H+) wird abgespalten. Umgekehrt gibt das NAD(P)H-Molekül ein Hydridion H– zusammen mit einem H+ aus der Umgebung ab, um 2 H-Atome zu übertragen.
| ⇓ | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||
| 2 H+, 2 e–
|
2 H+, 2 e–
|
Dehydrogenasen | 1.1.1.- | Ox | ||||||||
|
||||||||||||

Alternative Darstellung
| ⇓ | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||||
| 2 H+, 2 e–
|
2 H+, 2 e–
|
Dehydrogenasen | 1.1.1.- | Ox | ||||||||
|
||||||||||||


Idem für NADP+



- Weiterhin spielt Niacin eine Rolle bei der ADP-Ribosylierung, in dem es den ADP-Ribose-Rest liefert. Einige bakterielle Toxine sind ADP-Ribosyltransferasen, z.B. das Choleratoxin.
- Weiterhin ist NAD+ der Lieferant von cyclo-ADP-Ribose, einem Aktivator des muskulären Ryanodinrezeptors.
Pathobiochemie
BearbeitenLeichtere Niacin-Mangelerkrankungen sind unspezifisch: Reizbarkeit, Appetitlosigkeit, Konzentrations- und Schlafstörungen. Vollbild ist die Pellagra (pelle agra: rauhe Haut), sich durch die vier D auszeichnet: Diarrhoe, Dermatitis, Depression, Demenz. Ursachen sind einseitige Ernährung (Mais) oder Alkoholismus. Hypervitaminosen sind auch in hohen Dosen selten, Symptome sind Vasodilatation mit Hitzegefühl (Flush), Hauterscheinungen (Pruritus, Exanthem), Magen-Darm-Probleme, Leberschädigung.
Literatur
Bearbeiten- Magni G, Di Stefano M, Orsomando G, Raffaelli N, Ruggieri S. “NAD(P) biosynthesis enzymes as potential targets for selective drug design”. Curr. Med. Chem., 16:1372–90, 2009. PMID 19355893.
- Magni G, Amici A, Emanuelli M, Raffaelli N, Ruggieri S. “Enzymology of NAD+ synthesis”. Adv. Enzymol. Relat. Areas Mol. Biol., 73:135–82, xi, 1999. PMID 10218108.
- Magni G, Orsomando G, Raffelli N, Ruggieri S. “Enzymology of mammalian NAD metabolism in health and disease”. Front. Biosci., 13:6135–54, 2008. PMID 18508649.
- Ciampricotti R, el Gamal MI. “Unstable angina, myocardial infarction and sudden death after an exercise stress test”. Int. J. Cardiol., 24:211–8, August 1989. PMID 2504674.
- Bogan KL, Brenner C. “Nicotinic acid, nicotinamide, and nicotinamide riboside: a molecular evaluation of NAD+ precursor vitamins in human nutrition”. Annu. Rev. Nutr., 28:115–30, 2008. DOI:10.1146/annurev.nutr.28.061807.155443. PMID 18429699.
Weblinks
Bearbeiten- KEGG: Nicotinate and nicotinamide metabolism - Homo sapiens (human)
- reactome.org: Nicotinate metabolism
Pantothenat-Stoffwechsel
BearbeitenAllgemeines
BearbeitenPantothensäure ist als Bestandteil von Coenzym A im ganzen Stoffwechsel aktiv. Weiterhin findet man sie als funktionstragendes Element in der Fettsäuresynthase.
Biosynthese / Herkunft
Bearbeiten-Pantothenat.svg) |
 |
(R)-Pantothensäure bzw. (R)-Pantothenat (παντόθεν, pantothen (gr.): überall vorhanden) besteht aus β-Alanin und (R)-Pantoinsäure ((R)-Pantoat, 2,4-Dihydroxy-3,3-dimethyl-Butansäure). Das wasserlösliche Vitamin wird von Bakterien, Pilzen und Pflanzen produziert und muss mit der Nahrung zugeführt werden.
Biosynthese von 4'-Phosphopanthein und Coenzym A aus (R)-Pantothenat
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|||||||||||
| ATP
ADP |
Pantothenat-Kinase | 2.7.1.33 | Tr | Hallervorden-Spatz-S., HARP-S. | |||||||
|
|||||||||||
| Cystein, ATP
AMP, PPi |
Phosphopantothenat-- Cystein-Ligase | 6.3.2.5 | Lig | ||||||||
|
|||||||||||
|
|
FMN | Phosphopantothenoyl- cystein-Decarboxylase | 4.1.1.36 | Ly | |||||||
|
|||||||||||
| ATP
PPi |
1. |
AMP
H2O |
1) Pantethein-phosphat- Adenylyltransferase | 2.7.7.3 | Tr | ||||||
| 2) Nucleotid- Diphosphatase | 3.6.1.9 | Hyd | |||||||||
|
|||||||||||
| ATP
ADP |
Dephospho-CoA-Kinase | 2.7.1.24 | Tr | ||||||||
|
|||||||||||
-4%27-Phosphopantothenat.svg)
-4%27-Phosphopantothenoyl-L-cystein.svg)



Biologische Funktionen
Bearbeiten- Pantothensäure ist ein Bestandteil von Coenzym A (Struktur: 3'-Phospho-ADP - Pantothensäure - Cysteamin). Dieses aktiviert zahlreiche Metabolite über die energiereiche Bindung an seine SH-Gruppe als Thioester. Coenzym A (CoA-SH) ist von zentraler Bedeutung für den gesamten Stoffwechsel. Bsp.:
- Acetyl-CoA: Ethanol-Abbau, Dehydrierende Decarboxylierung von Pyruvat, Citratzyklus, Abbau von Fettsäuren, Biosynthese von Fettsäuren, Bildung von Acetylcholin, Ketonkörper-Biosynthese, Cholesterin-Biosynthese; Abbau ketogener Aminosäuren: Threonin-Abbau (Acetaldehyd), Lysin-Abbau, ((Phenylalanin- und) Tyrosin-Abbau (Acetoacetat), Biosynthese von Melatonin, Abbau von Leucin (HMG-CoA) und Isoleucin. Weiterhin: Biosynthese von N-Acetylglutamat, Acetylierung von Arzneimitteln in der Leber (Biotransformation)
- Propionyl-CoA: Abbau ungeradzahliger Fettsäuren, Alternativer Abbau von Threonin, Methionin-Abbau, Abbau von Valin und Isoleucin
- Succinyl-CoA: Citratzyklus, Häm-Biosynthese
- Acyl-CoA: Auf- und Abbau von Triacylgycerinen, Phosphoglyceriden, Sphingolipiden und Derivate.
- 4'-Phosphopantethein bildet die prostetische Gruppe des Acyl-Carrier-Proteins (ACP), seine Sulfhydryl-Gruppe bildet die zentrale SH-Gruppe der Fettsäuresynthase (Biosynthese gesättigter Fettsäuren).
- Pantothensäure ist auch an der Wundheilung (Granulationsgewebe) beteiligt.
Pathobiochemie
BearbeitenMangelerkrankungen sind selten und kommen z.B. bei chronisch-entzündlichen Magen-Darm-Erkrankungen oder Alkoholismus vor. „Burning-foot“-Syndrom, unspezifische Symptome.
Weblinks
Bearbeiten
Pyridoxalphosphat-Stoffwechsel
BearbeitenAllgemeines
BearbeitenPyridoxalphosphat ist das zentrale Vitamin des Aminosäurenstoffwechsels. Es ist z.B. als Cofaktor der Transaminasen und Decarboxylasen aktiv.
Biosynthese / Herkunft
Bearbeiten
Zur Vitamin B6-Gruppe (Pyridoxine) gehören das Pyridoxamin, das Pyridoxal und das Pyridoxin sowie die jeweiligen 5'-Phosphat-Ester und zusätzlich noch das Pyridoxat, das mit dem Urin ausgeschieden wird. Die wasserlöslichen Vitamine können bis auf Pyridoxat ineinander umgewandelt werden. Biologisch aktiv ist das Pyridoxal-5'-phosphat (PLP, PALP). Die Biosynthese erfolgt bei Bakterien, Pilzen und Pflanzen z.B. aus Glycerinaldehyd-3-phosphat (Glycolyse) und D-Ribulose-5-phosphat (Pentosephosphatweg) oder aus D-Erythrose-4-phosphat (Pentosephosphatweg).
Aktivierung der B6-Vitamine (Recycling von Pyridoxalphosphat)
Bearbeiten| ⇓ | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| ATP
ADP |
Pyridoxalkinase | 2.7.1.35 | Tr | |||||||
|
||||||||||


| ⇓ | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| ATP
ADP |
Pyridoxalkinase | 2.7.1.35 | Tr | |||||||
|
||||||||||
| O2
H2O2 |
Pyridoxinphosphat-Oxidase | 1.4.3.5 | Ox | PNPO-Defizienz | ||||||
|
||||||||||


| ⇓ | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| ATP
ADP |
Pyridoxalkinase | 2.7.1.35 | Tr | |||||||
|
||||||||||
| H2O, O2
NH3, H2O2 |
Pyridoxinphosphat-Oxidase | 1.4.3.5 | Ox | PNPO-Defizienz | ||||||
|
||||||||||


Biologische Funktionen
BearbeitenPyridoxalphosphat (PLP, PALP) ist das zentrale Vitamin des Aminosäurenstoffwechsels. Je nach Proteinanteil (Apo-Enzym) werden folgende Reaktionen katalysiert:
- Transaminierungen durch Transaminasen z.B. Alanin-Transaminase, Aspartat-Transaminase (Aspartatzyklus), Ornithin-Aminotransferase (Ornithinsynthese aus und -abbau zu Glutamat), Phosphoserin-Transaminase (Biosynthese von L-Serin und Glycin), Alanin--Glyoxylat-Transaminase, α-Aminoadipat-Transaminase (Lysin-Abbau), Tyrosin-Transaminase und Verzweigtkettige-Aminosäuren-Transaminase.
- Decarboxylierungen: Synthese biogener Amine wie z.B. GABA aus Glutamat, Tyramin aus Tyrosin und Dopamin aus L-Dopa, Histamin aus Histidin, Serotonin aus 5-Hydroxy-Tryptophan. Weiterhin Decarboxylierung von Phosphatidylserin zu Phosphatidylethanolamin und Biosynthese von Putrescin.
- Aldolspaltung: Aldolasen spalten Serin in Glycin und eine Hydroxymethylgruppe sowie Threonin in Glycin und Acetaldehyd.
- α-,β-Eliminierung: Die L-Serin-Deaminase (syn. L-Serin-Dehydratase) deaminiert Serin zu Pyruvat, die Threonin-Ammoniak-Lyase (syn. L-Threonin-Dehydratase) deaminiert Threonin.
- Transsulfurierung und Cystein-Biosynthese im Abbauweg des Methionins.
- Erster Schritt der Häm-Biosynthese als Cofaktor der δ-Aminolävulinat-Synthase.
- PLP ist außerdem ein Bestandteil der Glycogen-Phosphorylase, wobei PLP hier etwas anders arbeitet als im Aminosäurenstoffwechsel (Säure-Basen-Katalyse durch die Phosphat-Gruppe).
Funktionsweise (Reaktionen mit Aminosäuren)
BearbeitenDie Aldehyd-Gruppe von PLP bildet mit der Aminogruppe der Aminosäure eine Schiff-Base (Aldimin), unterstützt von einer kationischen Gruppe des Enzyms. Der PLP-Stickstoff zieht Elektronen an und schwächt dadurch die Bindungen am Cα-Atom der Aminosäure, so dass je nach Apo-Enzym unterschiedliche Reaktionen ermöglicht werden.
- Schwächung der Bindung zwischen dem Cα-Atom und der Aminogruppe: Transaminierung (PLP deaminiert eine Aminosäure und überträgt die Aminogruppe dann auf eine Ketosäure)
- Schwächung der Bindung zwischen dem Cα-Atom und der Carboxylgruppe: Decarboxylierung
- Schwächung der Bindung zwischen dem Cα- und dem Cβ-Atom: Aldolspaltung
- Schwächung der Bindung zwischen dem Cα- und dem H-Atom, sowie dem Cβ-Atom und einem Substituenten: α-,β-Eliminierung
Pathobiochemie
BearbeitenVitamin B6-Mangelerkrankungen sind selten und oft unspezifisch: Seborrhoische Dermatitis, Glossitis, neurologische Symptome (Krampfanfälle, Neuritis, Depressionen, Reizbarkeit), Wachstumsstörungen, Anämie.
Weblinks
Bearbeiten
Biotin-Stoffwechsel
BearbeitenBiosynthese / Herkunft
Bearbeiten
Mikroorganismen und Pflanzen bilden Biotin (Vitamin H) aus Pimelat, einer Heptadicarbonsäure.
Biologische Funktionen
BearbeitenBiotin (Vitamin H) ist das Coenzym zahlreicher Carboxylierungsreaktionen und über einen Lysin-Rest an das Apo-Enzym gebunden. Biotin bindet CO2 (HCO3-) und überträgt es unter ATP-Verbrauch auf das Empfängermolekül, z.B. in folgenden Reaktionen:
- Die Acetyl-CoA-Carboxylase carboxyliert Acetyl-CoA zu Malonyl-CoA, dem Substrat der Fettsäuresynthese
- Die Pyruvatcarboxylase carboxyliert im Mitochondrium Pyruvat zu Oxalacetat. Die ATP-abhängige Reaktion ist der erste Schritt der Gluconeogenese und zählt weiterhin zu den anaplerotischen Reaktionen, mit denen der Citratzyklus aufgefüllt werden kann.
- Die Propionyl-CoA-Carboxylase carboxyliert Propionyl-CoA zu D-Methylmalonyl-CoA, die erste Reaktion zum Abbau von Propionyl-CoA über Succinyl-CoA, das hernach in den Citratzyklus fließen kann. Propionyl-CoA fällt z.B. an beim Abbau von ungeradzahligen Fettsäuren, beim alternativen Abbau von Threonin, beim Methionin-Abbau, beim Abbau von Valin und Isoleucin sowie bei der Gallensäuren-Biosynthese.
- Die Methylcrotonyl-CoA-Carboxylase katalysiert einen Stoffwechselschritt im Abbau der verzweigtkettigen Aminosäure Leucin.
Abspaltung und Einbau von Bioptin
Bearbeiten| ⇓ | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. |
|---|---|---|---|---|---|---|---|
|
|||||||
| H20 | Biotinidase | 3.5.1.12 | Hyd | Biotinidase-Mangel | |||
|
|||||||
| Apo-[Carboxylase], ATP
AMP + PPi |
Holocarboxylase-Synthetase | 6.3.4.9, 6.3.4.10, 6.3.4.11, 6.3.4.15 | Lig | Holocarboxylase-Synthetase-Defizienz | |||
|
|||||||
| 2 H2O
2 Peptide |
Peptid-Hydrolase | 3.4.-.- | Hyd | ||||
|
|||||||
Pathobiochemie
BearbeitenBiotin-Mangelerkrankungen sind sehr selten. Hautentzündungen, Appetitlosigkeit, Parästhesien und Gliederschmerzen sowie psychiatrische Störungen kommen vor. Mögliche Ursachen sind eine Reduktion der Darmflora durch Antibiotika plus Biotin-arme Ernährung oder ein angeborener Biotinidase-Mangel.
Weblinks
Bearbeiten
Folat-Stoffwechsel
BearbeitenAllgemeines
BearbeitenDie biologisch aktive Form der Folsäure, die Tetrahydrofolsäure (THF), spielt als Lieferant von Ein-Kohlenstoff-Resten eine wichtige Rolle bei der Synthese von DNA- und RNA-Bausteinen. Zusammen mit Cobalamin (Vitamin B12) ist THF auch an der Remethylierung von Homocystein zu Methionin beteiligt. Folsäure- und Cobalamin-Mangel führen durch Störung der Zellteilung u.a. zur hyperchromatischen makrozytären Anämie.
Übersicht
BearbeitenHier sehen Sie eine Übersicht über den Folatstoffwechsel. Wichtige Schnittstellen mit anderen Stoffwechselwegen sind farblich markiert.
|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Einzelreaktionen
Bearbeiten
|
|||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
|
| ||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
|
|
|
| ||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||
|
| ||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
| ||||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||||||







 |
 |
 |
 |
 |
 |
Biosynthese und Chemie
BearbeitenFolsäure (folium (lat.): Blatt) wird von Mikroorganismen und Pflanzen gebildet, für Tiere ist es essentiell. Die Biosynthese von Folat und dem verwandten Biopterin erfolgt ausgehend vom Purin GTP, wobei das Purin-Ringsystem (2 Heterozyklen: Pyrimidin + Imidazol) in ein Pteridin-Ringsystem (2 Heterozyklen: Pyrimidin + Pyrazin) umgewandelt wird. Das vom GTP abgeleitete Pteridin heißt Pterin. Das fertige Folat-Molekül besteht aus Pterin, para-Aminobenzoesäure und Glutamat.
Biologische Funktionen
BearbeitenDie biologisch aktive Folsäure ist die Tetrahydrofolsäure (THF), die durch die Dihydrofolat-Reduktase (DHFR) aus Folsäure oder Dihydrofolsäure (DHF) gebildet wird.
Tetrahydrofolsäure ist als C1-Donator von Methyl- und Formylresten an zahlreichen Stoffwechselschritten beteiligt. Der Kohlenstoff stammt dabei meist aus der Aminosäure Serin und liefert N5,N10-Methylen-THF. Eine weitere C1-Quelle ist das Forminoglutamat aus dem Histidin-Abbau, das N5-Formimino-THF liefert, welches zu N5,N10-Methenyl-THF desaminiert wird. Weiterhin kann THF Formiat (Ameisensäure) binden in Form von N10-Formyl-THF.
Bedeutungen als C1-Donator:
- N10-Formyl-THF und N5,N10-Methenyl-THF: Purinbiosynthese (C-Atom 2 und 8) von AMP und GMP, wichtig für die DNA- und RNA-Synthese. Übertragen werden Formyl-Gruppen.
- N5,N10-Methylen-THF: Synthese des Pyrimidins Thymidylat (dTMP) aus dUMP, das für die DNA-Synthese benötigt wird. Übertragen werden Methyl-Gruppen.
- N10-Formyl-THF: Das Startcodon der Proteinbiosynthese ist das Codon AUG, welches gleichzeitig für Methionin kodiert. Der Initiationskomplex am Startcodon bindet bei Bakterien und in Mitochondrien (!) jedoch keine Methionin-beladene tRNA (Met-tRNAMet), sondern eine N-Formyl-methionin-tRNAfMet. (Die tRNAfMet ist nicht mit der tRNAMet identisch, beide werden jedoch von der gleichen Aminoacyl-tRNA-Synthetase beladen.) Nach der Bindung von Methionin an die tRNAfMet wird dieses formyliert. Während der Translation erfolgt meist schon die Deformylierung des Methionins, Methionin und weitere Aminosäuren können auch posttranslational durch Aminopeptidasen entfernt werden.
- N5-Methyl-THF: Unter Beteiligung von Vitamin B12 katalysiert die Methionin-Synthase die Remethylierung von Homocystein zu Methionin. Die Methylgruppe stammt dabei aus der N5-Methyl-THF. Methionin reagiert enzymkatalysiert mit ATP unter Abspaltung von drei Phosphat zu S-Adenosylmethionin, dem wichtigsten Methylgruppenüberträger im Intermediärstoffwechsel.
Pathobiochemie
BearbeitenEin Folat-Mangel führt über die Hemmung der DNA-Synthese zu Zellteilungsstörungen (Megaloblastäre Anämie), Diarrhoe, Gewichtsverlust und in der Frühschwangerschaft zu Spina bifida (auch assoziiert mit Störungen des Folat-Stoffwechsels, siehe OMIM: folate-sensitive neural tube defects) und Lippenspalte mit und ohne Gaumenspalte. Ursachen sind Mangelernährung (bes. Alkoholiker), erhöhter Bedarf (Schwangerschaft, Stillzeit, Hämolyse, Intrinsic-Faktor-Mangel), Malabsorption, Medikamente wie Antiepileptika (Phenytoin, Barbiturate, Primidon), Folsäureantagonisten (Methotrexat, Pyrimethamin, Trimethoprim) oder orale Kontrazeptiva (Resorptionshemmung). Eine weitere, häufige Ursache für Folatmangel ist das Vorliegen eines MTHFR Polymorphismus. Dieser häufige Gendefekt vermindert die Fähigkeit des Körpers Folsäure in das aktive Folat umzuwandeln. Dabei spielen besonders die SNP's 677 und 1298 eine große Rolle. In heterozygoter Form ist die Produktion des MTHFR Enzyms um 30% vermindert. In homozygoter oder compound heterozygoter Variante sogar bis zu 70 %. Da eine Störung des Folatmetabolismus auch die Homocystein Umwandlung zu Methionin beeinflusst existiert ein ausgeprägtes Risiko für thrombozytische Ereignisse und Gerinnungsstörungen. Das ist vor allem für den Erhalt einer Schwangerschaft evident. Bei mehrmaligen Aborten sollte daher unbedingt ein MTHFR Gentest durchgeführt werden.
Auch ein Mangel an Vitamin B12 kann wegen der B12/Folat-Schnittstelle bei der Methionin-Remethylierung zu einem funktionellen Folat-Mangel führen. Im Plasma liegt Folsäure überwiegend als 5-Methyl-THF (Monoglutamatform) vor. Nachdem es von der Zelle aufgenommen wurde muss es von der Methionin-Synthase in THF (Polyglutamatform) umgewandelt werden, um für die verschiedenen Reaktionen zur Verfügung zu stehen. Bei einem Cobalamin-Mangel ist dies gestört, 5-Methyl-THF wird so zur Sackgasse und THF fehlt.
Pharmakologie
BearbeitenStörungen der Zellvermehrung durch Nukleotid-Mangel tangieren v.a. proliferationsfreudige Gewebe wie Tumorzellen und das Immunsystem. Folatantagonisten wie Aminopterin und Methotrexat (MTX) können daher zur Immunsuppression bei Autoimmunerkrankungen und zur Wachstumshemmung bei Krebserkrankungen angewendet werden. MTX hemmt dabei insbesondere die Dihydrofolat-Reduktase (DHFR).
Da auch Mikroorganismen Folat benötigen können Dihydrofolat-Reduktase-Hemmer wie Trimethoprim und Pyrimethamin auch als Antibiotika eingesetzt werden. Diese werden meist mit Analoga der para-Aminobenzoesäure (Sulfonamide wie Sulfamethoxazol) kombiniert, die die mikrobielle Dihydropteroat-Synthase (EC 2.5.1.15) und damit gezielt die mikrobielle Folsäuresynthese hemmen. Ein solches Kombi-Präparat ist z.B. Cotrimoxazol.
Literatur
Bearbeiten- Whitehead VM. “Acquired and inherited disorders of cobalamin and folate in children”. Br. J. Haematol., 134:125–36, July 2006. DOI:10.1111/j.1365-2141.2006.06133.x. PMID 16846473.
Weblinks
Bearbeiten- KEGG: Folate biosynthesis - Homo sapiens (human)
- KEGG: One carbon pool by folate - Homo sapiens (human)
- RCSB PDB: Dihydrofolate Reductase
Cobalamin-Stoffwechsel
Bearbeiten |
|
Biosynthese / Herkunft
BearbeitenDer Biosynthese-Weg der Cobalamine zweigt vom Häm-Syntheseweg (Uroporphyrinogen III) ab. Das wasserlösliche Vitamin wird nur von Bakterien und Archaeen gebildet und besteht aus einem Corrin-Ringsystem, das zentral ein Kobaltion komplexiert. Anders als beim Häm-Molekül ist das Ringsystem nicht durchgehend Mesomerie-stabilisiert, d.h. die konjugierten Doppelbindungen sind nicht rundum vorhanden.
Die Resorption im terminalen Ileum durch Rezeptor-vermittelte Endozytose erfordert die Bindung von Vitamin B12 an das Glycoprotein intrinsic factor, der von den Belegzellen des Magens gebildet wird. Vitamin B12 wird im Blut von einem spezifischen Transportprotein befördert und in der Leber gespeichert. Da der Bedarf sehr gering ist kann es bei einer Unterversorgung mehrere Monate bis Jahre dauern, bis Mangelsymptome eintreten.
Chemische Formen
BearbeitenDie 6. Koordinationsstelle des Kobalt-Ions kann verschieden besetzt sein, so dass die verschiedenen Cobalamine resultieren, die auch ineinander umgewandelt werden können:
- Hydroxycobalamin - R: Hydroxyl-Gruppe (-OH)
- Adenosylcobalamin - R: Adenosin
- Desoxyadenosylcobalamin - R: Deoxyadenosin
- Cyanocobalamin - R: Cyanid (-C≡N)
- Methylcobalamin - R: Methyl-Gruppe (-CH3)
Biologische Funktionen
Bearbeiten- (5'-Desoxy)Adenosylcobalamin (AdoCbl) findet sich in den Mitochondrien als Cofaktor der Methylmalonyl-CoA-Mutase, die für die Umlagerung von Alkylresten sorgt, z.B. die Isomerisierung von Methylmalonyl-CoA zu Succinyl-CoA beim Abbau von Propionyl-CoA.
- Methylcobalamin (MeCbl) ist im Zytosol zu finden als Cofaktor der Methionin-Synthase, die die Remethylierung von Homocystein zu Methionin katalysiert (und damit auch die Regeneration des Methylgruppenüberträgers S-Adenosylmethionin), wobei die Methylgruppe von der N5-Methyl-THF stammt (Regeneration von THF).
Stoffwechselwege im Detail
BearbeitenBildung von Cobalamin II
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| ? | ||||||||||
|
||||||||||
| LMBRD1 (lysosomal cobalamin exporter) | MMA und Homocystinurie, Typ cblF | |||||||||
|
||||||||||
| ?
? |
MMACHC | MMA und Homocystinurie, Typ cblC | ||||||||
|
||||||||||
| ?
? |
MMADHC (C2orf25) | MMA und Homocystinurie, Typ cblD | ||||||||
|
||||||||||
Bildung von Adenosylcobalamin (AdoCbl) aus Cbl II
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| ?
? |
MMADHC (C2orf25) | MMA, Typ cblD, Variante 2 | ||||||||
|
||||||||||
| ?
? |
MMAB (Cob(I)alamin-Transferase) | Tr | MMA, Typ cblB | |||||||
|
||||||||||
| ?
? |
MMAA | MMA, Typ cblA | ||||||||
|
||||||||||
Bildung von Methylcobalamin (MeCbl) aus Cbl II
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| ?
? |
MMADHC (C2orf25) | Homocysteinurie, Typ cblD, Variante 1 | ||||||||
|
||||||||||
| NADPH/H+, 2 SAM | FAD, FMN | Methionin-Synthase-Reduktase | 1.16.1.8 | Ox | Homocysteinurie, Typ cblE | |||||
|
||||||||||
Die Methionin-Synthase-Reduktase regeneriert das spontan oxidierte Cob(II)alamin in der Methionin-Synthase mittels einer reduktiven Methylierung mit Hilfe von S-Adenosylmethionin (SAM) und NADPH/H+, da nur das reduzierte Methylcob(I)alamin als Kofaktor wirken kann.
Pathobiochemie
BearbeitenB12-Mangel
BearbeitenUrsachen:
- Intrinsic-Faktor-Mangel bei: Z.n. Gastrektomie, chronische Typ-A-Gastritis, Malabsorptionssyndrom (z.B. Sprue), Kurzdarmsyndrom, Fischbandwurmbefall.
- Dünndarm-Fehlbesiedlungen (insbesondere Störungen im Ileum).
- Hereditäre Störungen.
- Höchst selten bei einseitiger Ernährung.
Pathogenese:
- Durch die Schnittstelle von Folsäure und Cobalamin im Stoffwechselschritt der Methionin-Synthese, kommt es bei einem Mangel an Cobalamin auch zu einer mangelhaften Rückgewinnung von THF aus N5-Methyl-THF. Da für die Purinnukleotidsynthese (AMP, GMP) N10-Formyl-THF und für die dTMP-Synthese aus dUMP (Pyrimidine) N5,N10-Methylen-THF benötigt wird, kommt es nachfolgend zu Störungen der DNA- und RNA-Synthese. Besonders betroffen sind regenerationsfreudige Organe mit hoher Zellteilungsrate wie Knochenmark und Schleimhäute.
- Vitamin B12-Mangel führt ebenfalls über die B12/Folat-Schnittstelle auch zu einem Mangel an S-Adenosylmethionin, was neben der DNA/RNA-Synthesestörung zu den neurologischen Störungen führen soll.
- Das nicht isomerisierte Methylmalonyl-CoA wird verstärkt zu Methylmalonat hydrolysiert. Methylmalonsäure soll einen allgemeinen toxischen Effekt haben. Der Nachweis im Urin ist ein empfindlicher Indikator für Vitamin B12-Mangel.
Klinisches Krankheitsbild:
- Perniziöse Anämie (makrozytäre megaloblastäre Anämie), sensorische Neuropathie (20-30 %), Glossitis, Diarrhö.
Genetische Störungen des Cobalaminstoffwechsels
BearbeitenEnzymdefekte können die Bereitstellung von Cobalamin II - der gemeinsamen Vorstufe von Adenosylcobalamin und Methylcobalamin - oder isoliert einen der beiden Pfade betreffen. Dadurch werden die davon abhängigen Enzyme Methylmalonyl-CoA-Mutase und/oder Methionin-Synthase in ihrer Funktion beeinträchtigt mit der Folge einer Methylmalonylacidämie (durch gestörten Abbau von Propionyl-CoA) und/oder Homocysteinämie (durch Störung der Methionin-Rückgewinnung aus Homocystein).
Pharmakologie und Toxikologie
BearbeitenHydroxocobalamin eignet sich zur Behandlung von Cyanid-Vergiftungen.
Literatur
Bearbeiten- Manoli I, Venditti CP., et al.. “Methylmalonic Acidemia”. Gene Reviews,. PMID 20301409.
- Watkins D, Rosenblatt DS. “Inborn errors of cobalamin absorption and metabolism”. Am J Med Genet C Semin Med Genet, 157:33–44, February 2011. DOI:10.1002/ajmg.c.30288. PMID 21312325.
- Froese DS, Gravel RA. “Genetic disorders of vitamin B₁₂ metabolism: eight complementation groups--eight genes”. Expert Rev Mol Med, 12:e37, 2010. DOI:10.1017/S1462399410001651. PMID 21114891.
- Morel CF, Watkins D, Scott P, Rinaldo P, Rosenblatt DS. “Prenatal diagnosis for methylmalonic acidemia and inborn errors of vitamin B12 metabolism and transport”. Mol. Genet. Metab., 86:160–71, 2005. DOI:10.1016/j.ymgme.2005.07.018. PMID 16150626.
Weblinks
Bearbeiten
Ascorbat-Stoffwechsel
BearbeitenBiosynthese / Herkunft
BearbeitenPflanzen, Mikroorganismen und die meisten Tiere bilden sich Vitamin C (L-Ascorbinsäure, L-Ascorbat) aus Glucuronsäure. Einige Wirbeltiere, darunter Primaten und Meerschweinchen, sind dazu nicht mehr in der Lage.
Biochemische Funktionen und physiologische Bedeutung
BearbeitenVitamin C kann schrittweise Elektronen abgeben. Dadurch kann es als 1-Elektronendonator (Reduktionsmittel) wirken. Redoxgleichgewicht:
| ⇓ | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
|
|
L-Dehydroascorbinsäure
L-Semidehydroascorbinsäure |
Ox | ||||||||
|
||||||||||
|
|
Ox | |||||||||
|
||||||||||
| 2 e-, 2 H+
|
2 e-, 2 H+
|
Ox | ||||||||
|
||||||||||



Vitamin C ist an folgenden Reaktionen beteiligt:
- Antioxidans: Als Radikalfänger inaktiviert Ascorbinsäure radikale Sauerstoff- und Stickstoffspezies, indem es diesen Elektronen anbietet. Dadurch werden wichtige biologische Strukturen z.B. Membranlipide vor der Oxidation geschützt. Zwei Ascorbylradikale können dann zu Ascorbinsäure und Dehydroascorbinsäure dismutieren. Als Co-Antioxidans kooperiert es mit und regeneriert Tocopherol (Vitamin E), ebenfalls ein Radikalfänger.
- Monooxidase-Reaktionen:
- Die Dopamin-β-Monooxygenase hydroxyliert Dopamin zu Noradrenalin (Katecholaminbiosynthese)
- Dioxidase-Reaktionen:
- Kollagensynthese: Die Prolyl- (EC 1.14.11.2, EC 1.14.11.7) und die Lysyl-Hydroxylase (EC 114.11.4) hydroxylieren das Prolin und Lysin im Prokollagen. Vitamin C aktiviert die Hydroxylase durch Reduktion des Fe3+ zu Fe2+.
- Carnitin-Biosynthese: Die γ-Butyrobetain-Dioxygenase (EC 1.14.11.1) und die Trimethyllysin-Dioxygenase (EC 1.14.11.8) arbeiten mit Vitamin C (und Eisen).
- Synthese peptidischer Neurotransmitter bzw. Hormone: Die Peptidylglycin-Monooxygenase (Peptidylglycin-α-Amidierung-Monooxygenase) (EC 1.14.17.3) katalysiert Vitamin-C-abhängig die Amidierung am C-Ende von z.B. ADH, CRH, GRH, Bombesin, Calcitonin, Pankreozymin-Cholezystokinin, VIP, Gastrin u.a.m.
- Mischfunktionelle Monooxygenasen:
- Phase I-Transformation: Zusammen mit P450 ist Vitamin C an der Hydroxylierung von Steroiden, Xenobiotika und Gallensäuren beteiligt (?).
- Komplexbildung: Vitamin C ist ein Ligand für Metallionen. Durch die Bildung von Fe2+-Chelaten im Darm erhöht Vitamin C z.B. die Eisen-Resorption.
Recycling von Ascorbinsäure
BearbeitenAscorbinsäure wird nach ihrer Oxidation zu Semidehydroascorbinsäure und Dehydroascorbinsäure nicht notwendigerweise abgebaut. Es stehen mehrere Mechanismen bereit, um diese Stoffe zu Ascorbinsäure zu reduzieren. Dehydroascorbinsäure wird mithife von Glutathion und der Glutathiontransferase Omega-1/-2 recycliert:
| ⇓ | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| 2 GSH GSSG |
Glutathion-S-Transferase Omega (1/2) | 2.5.1.18 | Tr | |||||||
|
||||||||||
Auf der Zytosolseite der mitochondriellen Membran steht ein Komplex aus Cytochrom b5 und NADH-Cytochrom b5-Reduktase bereit, der im reduzierten Zustand (Fe(II)-Häm) die Reduktion von Semidehydroascorbat bewerkstelligt. Die Reduktase verbraucht im Folgenden NADH, um das Fe(III) des Cytochroms wieder in den Anfangszustand zu versetzen.
Pathobiochemie
BearbeitenMangelerkrankungen: Skorbut: Kapillarfragilität mit Hautblutungen (v.a. subperiostale Ekchymosen), Zahnfleischblutungen, Zahnausfall, Störungen des Knochen- und Zahnstoffwechsels, allgemeine Schwäche; Müller-Barlow-Erkrankung (Kleinkinder).
Hypervitaminosen: / (evtl. Oxalat-Nierensteine bei Megadosen, prooxidative Wirkung hoher Dosen nicht ausgeschlossen)
Weblinks
Bearbeiten
Retinol-Stoffwechsel
BearbeitenAllgemeines
BearbeitenVitamin A ist in Form von Retinal am Sehvorgang beteiligt und als Retinoat an der Genexpression.
Provitamin A (β-Caroten) und Vitamin A1 (Retinol)
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | ||
|---|---|---|---|---|---|---|---|---|---|
|
|||||||||
| O2
all-trans-Retinal |
Gallen- salze, Fe | Beta-Caroten-15,15'-Monooxygenase | 1.14.99.36 | Ox | |||||
|
|||||||||
| NADH/H+ | NADH/H+ | Retinol-Dehydrogenase | 1.1.1.105 | Ox | Fundus albipunc- tatus | ||||
|
|||||||||
| Palmitoyl-CoA
CoA-SH |
1. |
Palmitat
H2O |
1) Retinol-O-Fettsäuretransferase | 2.3.1.76 | Tr | ||||
| 2) Retinyl-palmitat-Esterase | 3.1.1.21 | Hyd | |||||||
| 2) All-trans-Retinyl-palmitat-Hydrolase | 3.1.1.64 | Hyd | |||||||
|
|||||||||


β-Carotin bzw. Provitamin A ist die pflanzliche Vorstufe der fettlöslichen Vitamine der A-Gruppe. All-trans-Retinol ist das eigentliche Vitamin A (Vitamin A1). Die Retinoide enthalten zahlreiche konjugierte Doppelbindungen und sie leiten sich, wie an den Isopren-Einheiten zu erkennen ist, aus den Anfangsstufen der Steroid-Biosynthese ab. Retinol wird im Dünndarm passiv oder carriervermittelt aufgenommen. Als Ester an CRBP (cellular retinol binding protein) gebunden erfolgt der Transport in Chylomikronen über die Darmlymphe in den Kreislauf. In der Leber wird CRBP abgespalten und Retinol als Retinylester (Retinyl-palmitat) in den Ito-Zellen (Perizyten im Disse'schen Raum) gespeichert. Die Ausscheidung erfolgt hepatobiliär nach der Oxidation von all-trans-Retinal zu all-trans-Retinoat und Glucuronidierung (s.u.).
Bildung von all-trans-Retinoat aus all-trans-Retinal
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | ||
|---|---|---|---|---|---|---|---|---|---|
|
|||||||||
| H2O, NAD+ | FAD | Retinal- Dehydrogenase | 1.2.1.36 | Ox | |||||
|
|||||||||
| UDP-D-Glucuronat
UDP |
FAD | Glucuronosyl- transferase | 2.4.1.17 | Tr | Hyperbilirubinämie I (Gilbert-S.), Crigler- Najjar-S. Typ I und II | ||||
|
|||||||||
Retinoat fungiert als Wachstumsfaktor, in dem es an verschiedene Subtypen von intrazellulären heteromeren Retinsäurerezeptoren (Retinoic acid receptor (RAR) und RXR) bindet, die als Transkriptionsfaktoren wirken. Darüber reguliert es die Zellproliferation und -differenzierung. Betroffen sind v.a. regenerationsfreudige Organe wie Schleimhäute, Haut und Blutbildung.
In den Geschlechtsorganen beeinflussen die Retinoide die Produktion von Testosteron, die Reproduktion und die fetale Entwicklung.
all-trans- und 11-cis-Retinal - die Schlüsselmoleküle des Sehvorgangs
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | ||
|---|---|---|---|---|---|---|---|---|---|
|
|||||||||
| Retinal-Isomerase
(Pigmentepithel) |
5.2.1.3 | Iso | |||||||
|
|||||||||
|
(Rezeptor-Außensegment) | ||||||||
|
|||||||||
|
hν ~ |
(Rezeptor-Außensegment) | ||||||||
|
|||||||||
| (Rezeptor-Außensegment) | |||||||||
|
|||||||||
| (Rezeptor-Außensegment) | |||||||||
|
|||||||||
|
(Rezeptor-Außensegment) | ||||||||
|
|||||||||



Die Aldehyd-Isomere 11-cis-Retinal und all-trans-Retinal sind für den Sehvorgang essentiell, indem sie die physikalische Energie eines Photons in eine chemische Isomerisierung übersetzen. Das reaktive 11-cis-Retinal bildet zusammen mit dem Glycoprotein (Scot)Opsin das Rhodopsin, das Sehpigment. Durch Lichteinwirkung wird 11-cis-Retinal zu all-trans-Retinal isomerisiert. Die Isomerisierung führt zu einer mehrstufigen Konformationsänderung des Proteinanteils bis zum Metarhodopsin II. Dieses aktiviert dann vielfach das G-Protein Transducin, welches sein GDP gegen GTP eintauscht und nun mit seiner α-Einheit eine Phosphodiesterase (PDE) bindet und aktiviert. Die PDE spaltet dann den von einer Guanylatcyclase gebildeten sekundären Botenstoff cGMP, der Na+-Ca2+-Kanäle in der Membran offen hält. Die Ionen-Kanäle schließen und - anders als bei vielen anderen Rezeptoren - kommt es nun zur Hyperpolarisation und Absinken der Transmitterfreisetzung, wodurch das Signal weitergeleitet wird.
All-trans-Retinal wird in das Pigmentepithel transportiert, dort wieder zum reaktionsfähigen 11-cis-Retinal isomerisiert und wieder in das Rezeptor-Außensegment gebracht.
Ein Rhodopsin-ähnliches Protein, das Bakteriorhodopsin findet sich in halophilen Archaeen, die das Pigment zur Photosynthese nutzen.
Bildung von 11-cis-Retinol aus 11-cis-Retinal
Bearbeiten| ⇓ | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | ||
|---|---|---|---|---|---|---|---|---|---|
|
|||||||||
| NADH/H+ | NADH/H+ | Retinol-Dehydrogenase | 1.1.1.105 | Ox | Fundus albipunctatus | ||||
|
|||||||||

Pathobiochemie
BearbeitenMangelerkrankungen: Am Auge: Sehstörungen (Nachtblindheit, erhöhte Blendempfindlichkeit), Keratomalazie, Xerophthalmie; Hyperkeratose der Talgdrüsen, Atrophie von Schleimhäuten und Speicheldrüsen.
Hypervitaminosen: Gastrointestinale (Übelkeit, Erbrechen) und neurologische Symptome (Schwindel, Kopfschmerzen), bei sehr hoher parenteraler Dosierung: Alopezie, Hepatosplenomegalie. Teratogen! (Bei höherer Dosierung müssen Retinoid-Präparate 2 Jahre vor der Empfängnis abgesetzt werden!) Erhöhung des Lungenkrebsrisikos bei Rauchern.
Pharmakologie
BearbeitenRetinoide werden bei schweren Fällen von Akne eingesetzt. Genutzt werden dabei die antikeratinisierende / komedolytische und die antiinflammatorische Wirkung. In der Schwangerschaft können Retinoide allerdings zu schweren Missbildungen führen. Auch nach dem Absetzen der fettlöslichen und damit lange im Körper verbleibenden Retinoide muss noch über mehrere Monate eine Schwangerschaft sicher verhütet werden.
Weblinks
Bearbeiten
Vitamin K-Stoffwechsel
BearbeitenDer Vitamin K-Zyklus
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| NAD(P)H/H+ | FAD | NAD(P)H-Dehydrogenase | 1.6.5.2 | Ox | ||||||
|
||||||||||
|
γ-Glutamyl-Carboxylase (GGCX) | 6.4.-.- | Lig | VKCFD1 | ||||||
|
||||||||||
|
Vitamin-K-Epoxid-Reduktase | 1.1.4.1 | Ox | VKCFD2 | ||||||
|
||||||||||







Allgemeines
Bearbeiten |
 |
 |
Vitamin K kommt in zwei Formen vor:
- Phyllochinon (Vitamin K1) - Menadion + Phytylseitenkette. Vorkommen: Pflanzen.
- Menachinon (Vitamin K2) - Menadion + Difarnesylrest mit variabler Anzahl an Isopreneinheiten. Vorkommen: Bakterien.
- Menadion (Vitamin K3) - Synthetisch. Keine medizinische Anwendung.
Vitamin K ist essentiell für die γ-Carboxylierung u.a. der Vorstufen der prokoagulatorischen Gerinnungsfaktoren II (Prothrombin), VII, IX und X sowie des antikoagulatorischen Protein C in der Leber. Diese posttranslationale Modifikation erhöht das Bindungsvermögen für Kalzium, das für die Blutgerinnung essentiell ist.
Klinischer Bezug
BearbeitenPharmakologie: Die Cumarine Warfarin und Phenprocoumon hemmen die Vitamin-K-Epoxid-Reduktase und dadurch die γ-Carboxylierung. Beeinträchtigt wird davon vor allem das extrinsische Gerinnungssystem. Laborchemisch ist der Quick-Wert erniedrigt bzw. die INR erhöht. Ausnutzen lässt sich dieser Effekt für die Rezidivprophylaxe von Thrombosen und zur Prophylaxe thromboembolischer Ereignisse bei Vorhofflimmern. Die wichtigste Nebenwirkung ist das erhöhte Risiko gefährlicher Blutungen. Der Effekt lässt sich durch die Gabe von Vitamin K antagonisieren, was einige Tage dauert. Vitamin K-reiche Lebensmittel z.B. Kohlgemüse können die Wirksamkeit ebenfalls abschwächen.
Toxikologie: Vergiftungen durch Cumarine (Medikamente oder Rodentizide (Rattengift) auf Cumarin-Basis) werden mit Vitamin K antagonisiert. Da die Neubildung der Gerinnungsfaktoren einige Tage dauert, müssen diese im Notfall auch direkt substituiert werden, z.B. mit dem nicht ganz billigen Blutprodukt PPSB.
Pathobiochemie: Bei der Leberzirrhose und beim akuten Leberversagen kommt es zu einem Abfall des Quicks mit erhöhter Blutungsneigung durch die beeinträchtigte Synthese von Gerinnungsfaktoren.
Beim Morbus haemorrhagicus neonatorum führt ein Vitamin K-Defizit in den ersten Lebenswochen zu Hirnblutungen. Aus diesem Grunde bekommen alle Neugeborenen eine Vitamin K-Prophylaxe im Rahmen der Vorsorgeuntersuchungen U1, U2 und U3.
Literatur
Bearbeiten- Silva PJ, Ramos MJ. “Reaction mechanism of the vitamin K-dependent glutamate carboxylase: a computational study”. J Phys Chem B, 111:12883–7, November 2007. DOI:10.1021/jp0738208. PMID 17935315.
Weblinks
BearbeitenUbichinon-Stoffwechsel
BearbeitenAllgemeines
BearbeitenUbichinon ist am Elektronentransport in der Atmungskette beteiligt.
Biosynthese von Ubichinon (Coenzym Q)
BearbeitenTeil 1.: Verlängerung der Polyprenyl-Kette
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| Isopentenyl-PP
PPi |
Farnesyltranstransferase | 2.5.1.29 | Tr | |||||||
|
||||||||||
| Isopentenyl-PP
PPi |
? | ? | Tr | |||||||
|
||||||||||
| Isopentenyl-PP
PPi |
Trans-Pentaprenyltranstransferase | 2.5.1.33 | Tr | |||||||
|
||||||||||
| Isopentenyl-PP
PPi |
Trans-Hexaprenyltranstransferase | 2.5.1.30 | Tr | |||||||
|
||||||||||
| Isopentenyl-PP
PPi |
Trans-Octaprenyltranstransferase | 2.5.1.11 | Tr | |||||||
|
||||||||||
| Isopentenyl-PP
PPi |
Trans-Octaprenyltranstransferase | 2.5.1.11 | Tr | |||||||
|
||||||||||
| Isopentenyl-PP
PPi |
? | 2.5.1.- | Tr | |||||||
|
||||||||||








Teil 2.: Verknüpfung mit 4-Hydroxybenzoat und Modifikation des Ring-Systems
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
|
Parahydroxybenzoat- Polyprenyltransferase Coq2 | 2.5.1.-
(2.5.1.39) |
Tr | |||||||
|
||||||||||
| (1/2 O2) ?
? |
? | ? | Ox | |||||||
|
||||||||||
| S-Adenosylmethionin | Hexaprenyldihydroxybenzoat- Methyltransferase Coq3 | 2.1.1.114 | Tr | |||||||
|
||||||||||
| ?
(CO2) ? |
? | ? | ? | |||||||
|
||||||||||
| O2
H2O |
Monooxygenase Coq6 | 1.14.13.- | Ox | |||||||
|
||||||||||
| S-Adenosylmethionin | Methyltransferase Coq5 | 2.1.1.- | Tr | |||||||
|
||||||||||
| O2, NADPH/H+
H2O, NADP+ |
Monooxygenase Coq7 | 1.14.13.- | Ox | |||||||
|
||||||||||
| S-Adenosylmethionin | Hexaprenyldihydroxybenzoat- Methyltransferase Coq3 | 2.1.1.114 | Tr | |||||||
|
||||||||||










Ubichinon (Coenzym Q, CoQ, Q) leitet sich - wie Vitamin K und Vitamin E - vom Farnesylpyrophosphat bzw. ganz allgemein von den Terpenen (Poly-Isoprenoide) ab. Deren Biosynthese ist im Kapitel Cholesterin-Biosynthese beschrieben. Die Länge der Isoprenoid-Kette ist Spezies-abhängig. Der Mensch produziert hauptsächlich Ubichinon mit 10 Isopren-Einheiten, das sog. Q10. Das Ubichinon aus der Nahrung besitzt 6 bis 10 Einheiten, wobei die Moleküle mit kürzerer Polyprenyl-Kette im Körper mit Isopren-Einheiten zum Q10 aufgestockt werden.
Biologische Funktionen
Bearbeiten| ⇓ | Subst. | ⇑ | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| e-
|
e-
|
Ox | ||||||||
|
||||||||||
| e-, 2 H+
|
e-, 2 H+
|
Ox | ||||||||
|
||||||||||


Ubichinon ist neben dem Cytochrom c das zweite mobile Elektronentransportmittel in der Atmungskette. Während Cytochrom c Elektronen vom Komplex III auf Komplex IV überträgt, leitet Q diese von den Dehydrogenase-Komplexen I und II auf Komplex III über.
Im Unterschied zum Cytochrom c müssen bei vollständiger Reduktion des Ubichinons zum Ubihydrochinon zwei Protonen zum Ladungsausgleich aufgenommen werden (dies entspricht formal, aber nicht mechanistisch einer Wasserstoffübertragung).
Weblinks
Bearbeiten
Biopterin-Stoffwechsel
BearbeitenAllgemeines
Bearbeiten
Biopterin ist ein nicht-essentieller Co-Faktor. Das Molekül enthält wie die Folsäure und das Flavin ein stickstoffhaltiges Pteridin-Ringsystem. Alle drei werden aus dem Purin GTP gebildet, davon jedoch nur das Biopterin auch im Menschen.
Das Dihydrobiopterin/Tetrahydrobiopterin-System bildet neben dem NAD- und FAD-System ein weiteres wichtiges Redox-System. Eine besondere Rolle spielt es bei der Oxidation (Hydroxylierung) aromatischer Ringe, wobei es molekularen Sauerstoff verwendet.
Vorkommen:
- Biosynthese von Serotonin aus Tryptophan
- Biosynthese von Tyrosin aus Phenylalanin
- Biosynthese von L-DOPA aus Tyrosin
Vom Biopterin-Syntheseweg zweigt auch die Bildung von Molybdopterin ab, einem Molybdän-bindenden Cofaktor.
Biosynthese von Biopterin aus GTP
Bearbeiten| ⇓ | Subst. | (⇑) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| H2O
|
GTP-Cyclohydrolase | 3.5.4.16 | Hyd | HPABH4B, DOPA- responsive Dystonie | ||||||
|
||||||||||
| H2O | GTP-Cyclohydrolase | 3.5.4.16 | Hyd | HPABH4B, DOPA- responsive Dystonie | ||||||
|
||||||||||
| GTP-Cyclohydrolase | 3.5.4.16 | Hyd | HPABH4B, DOPA- responsive Dystonie | |||||||
|
||||||||||
|
|
GTP-Cyclohydrolase | 3.5.4.16 | Hyd | HPABH4B, DOPA- responsive Dystonie | ||||||
|
||||||||||
|
|
Mg | 6-Pyruvoyltetra-hydropterin-Synthase | 4.2.3.12 | Ly | HPABH4A | |||||
|
||||||||||
| NADPH/H+ | Sepiapterin-Reduktase | 1.1.1.153 | Ox | L-Dopa- responsive Dystonie | ||||||
|
||||||||||
| NADPH/H+ | Sepiapterin-Reduktase | 1.1.1.153 | Ox | L-Dopa- responsive Dystonie | ||||||
|
||||||||||
| NAD(P)+ | NAD(P)+ | 6,7-Dihydropteridin-Reduktase | 1.5.1.34 | Ox | HPABH4C | |||||
|
||||||||||



-amino-4-oxopyrimidin.svg)
-dihydropteridintriphosphat.svg)




Funktionsweise des Dihydrobiopterin/Tetrahydrobiopterin-Systems
Bearbeiten| ⇓ | Subst. | (⇑) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| O2, Aromat
Hydroxy-Aromat |
Fe | Monooxygenase (Hydroxylase) | 1.14.16.1 1.14.16.2 1.14.16.4 | Ox | ||||||
|
||||||||||
|
|
4a-Hydroxytetrahydrobiopterin- Dehydratase | 4.2.1.96 | Ly | HPABH4D | ||||||
|
||||||||||
| NAD(P)H/H+ | NAD(P)H/H+ | 6,7-Dihydropteridin-Reduktase | 1.5.1.34 | Ox | HPABH4C | |||||
|
||||||||||

Die Hydroxylase überträgt vom molekularen Sauerstoff ein Sauerstoff-Atom auf den Aromaten und ein Sauerstoff-Atom auf BH4, dass dadurch zum 4a-Hydroxytetrahydrobiopterin oxidiert wird. Dieses gibt den aufgenommen Sauerstoff in Form von Wasser wieder ab. Der verlorene Wasserstoff wird durch die nachfolgende Reduktion wieder ersetzt.
Biosynthese von Molybdopterin
Bearbeiten| ⇓ | Subst. | (⇑) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| S-Adenosylmethionin, H2O
L-Methionin, Deoxyadenosin, PPi |
MOCS1 | ? | Cyc | MoCo-Defizienz Typ A | ||||||
|
||||||||||
| 2 L-Cystein, Cu
2 L-Alanin, H+ |
Molybdopterin-Synthase (MOCS2) | 2.8.1.- | Tr | MoCo-Defizienz Typ B | ||||||
|
||||||||||
| Molybdat, ATP, H2O
Cu, AMP, PPi |
Gephyrin | 2.-.-.- | Tr | MoCo-Defizienz Typ C | ||||||
|
||||||||||
| L-Cystein | MOCOS | 2.8.1.- | Tr | Xanthinurie II | ||||||
|
||||||||||




Molybdopterin bindet mit seinen zwei Thio-Gruppen ein Molybdän-Atom (mit zwei Sauerstoffatomen, also eigentlich MoO2) und dient in dieser Form als Cofaktor der Sulfit-Oxidase. Um die Enzyme Xanthin-Oxidase (XOD) und Aldehyd-Oxidase zu aktivieren, wird noch ein weiterer Schritt, der Austausch eines der o.g. Sauerstoffatome gegen Schwefel benötigt. Es exisieren also zwei verschiedene Molybdän-Cofaktoren im Menschen.
Der Molybdän-Cofaktor wird zu Urothion abgebaut.
Literatur
BearbeitenTetrahydrobiopterin: PMID 10727395
Molybdopterin: PMID 19675644 PMID 19623604 PMID 18092812 PMID 17351249 PMID 16261263 PMID 2522104
Weblinks
Bearbeiten
Liponsäure-Stoffwechsel
BearbeitenAllgemeines
BearbeitenLiponsäure ist als Cofaktor an der dehydrierenden Decarboxylierung von α-Ketosäuren und an der Spaltung von Glycin beteiligt.
Liponsäure wird unter Einbau von Schwefel aus der Fettsäure Oktanat gebildet
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| 2 S0, 2 SAM | Lipoyl-Synthase | 2.8.1.8 | Tr | |||||||
|
||||||||||
| Apoprotein
ACP |
Lipoyl(octanoyl)-Transferase | 2.3.1.181 | Tr | |||||||
|
||||||||||

Alternativer Syntheseweg durch umgekehrte Reihenfolge:
| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| Apoprotein
ACP |
Lipoyl(octanoyl)-Transferase | 2.3.1.181 | Tr | |||||||
|
||||||||||
| 2 S0, 2 SAM | Lipoyl-Synthase | 2.8.1.8 | Tr | |||||||
|
||||||||||

Der an den Lysyl-Rest eines Proteins gebundende Liponsäure-Rest kann leicht oxidiert und reduziert werden, bildet also ein Redox-System. Die Reduktion erfolgt durch die temporäre Bindung und Abspaltung anderer Moleküle an eines der Schwefelatome. Der dabei gebildete reduzierte Dihydroliponsäure-Rest kann unter Gewinn von NADH/H+ reoxidiert werden. Protein-Lipoyllysin ist an folgenden Reaktionen beteiligt:
- Als Bestandteil der E2-Untereinheit der drei Ketosäure-Dehydrogenase-Komplexe an der dehydrierenden Decarboxylierung von α-Ketosäuren.
- Als Bestandteil des Glycin cleavage-Systems (Protein H) Abbau von Glycin.
Weblinks
Bearbeiten
Eisen
BearbeitenAllgemeines
BearbeitenIm Körper befinden sich etwa 3 bis 5 g Eisen, etwa 2/3 davon im Hämoglobin, wo es im Häm für die Sauerstoffbindung verantwortlich ist. Daneben findet sich Eisen in zahllosen Enzymen, von denen viele an Elektronenübertragungen (Redoxreaktionen) beteiligt sind.
Funktion
Bearbeiten| Eisen | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
 | ||||||||||
| Basisdaten und Verweise | ||||||||||
|
Eisen-Schwefel-Zentren finden sich in:
- Aconitase - Citratzyklus
- Eisensensorisches Protein
- NADH-Dehydrogenase - Atmungskette (Komplex I)
- Succinat-Dehydrogenase - Atmungskette (Komplex II)/Citratzyklus
- Cytochrom-c-Reduktase - Atmungskette (Komplex III)
- Xanthin-Oxidase (XOD) - Harnsäure-Abbau
Eisen findet sich als Zentralion im Häm. In dieser Form dient es folgenden Proteinen als Cofaktor:
- Hämoglobin
- Myoglobin
- NAD(P)H-Oxidase - ROS-Bildung
- Katalase - siehe unter Atmungskette
- Succinat-Dehydrogenase - Atmungskette (Komplex II)/Citratzyklus
- Cytochrom-c-Reduktase - Atmungskette (Komplex III)
- Cytochrom c - Atmungskette
- Cytochrom-c-Oxidase - Atmungskette (Komplex IV)
- Cytochrom-c-Peroxidase
- Cyclooxygenase (COX) - Arachidonsäure-Stoffwechsel (Hinweis: Eisen bzw. Häm wird weder bei KEGG noch bei EC->PDB als Cofaktor genannt, aber z.B. bei PDB.)
- Lactoperoxidase - Phenylalanin-Abbau
- Thyreoperoxidase - Schilddrüsenhormon-Synthese
- Aldehyd-Oxidase - Serotoninabbauweg
- Tryptophan-2,3-Dioxygenase - Tryptophan-Abbau
- Indolamin-2,3-Dioxygenase - Tryptophan-Abbau
- Sulfit-Oxidase - Schwefel-Stoffwechsel
In Form von Häm-Thiolat findet man das Häm als Cofaktor der Cytochrom P450-Proteine (CYP), die für die Biotransformation Phase I verantwortlich sind. Dazu gehören folgende Enzyme:
- Thromboxan-A-Synthase (CYP5A) - Biosynthese von TXA2
- Unspezifische Monooxygenase (CYP1A1, CYP1A2, CYP1B1, CYP2A, CYP2B, CYP2C, CYP2D, CYP2E, CYP2F, CYP2J, CYP2S, CYP3A, CYP4B, CYP4X, CYP4Z, CYP19A) - Steroidhormon-Stoffwechsel
- Prostacyclin-Synthase (CYP8A) - Biosynthese von Prostacyclin
- Sterol-14-Demethylase (CYP51A) - Cholesterin-Biosynthese
- Calcidiol-1-Monooxygenase (CYP27B) - Calcitriol-Bildung
- Cholesterol-7α-Monooxygenase (CYP7A1) - Gallensäuren-Biosynthese
- Cholestanetriol-26-Monooxygenase (CYP27A) - Gallensäuren-Biosynthese
- Cholesterol-Monooxygenase (20-22-Desmolase, CYP11A) - Steroidhormon-Stoffwechsel
- Steroid-11β-Monooxygenase (11β-Hydroxylase, CYP11B) - Steroidhormon-Stoffwechsel
- Corticosteron-18-Monooxygenase (18-Hydroxylase, CYP11B) - Steroidhormon-Stoffwechsel
- Steroid-17α-Monooxygenase (17α-Hydroxylase, CYP17A) - Steroidhormon-Stoffwechsel
- Steroid-21-Monooxygenase (21-Hydroxylase, CYP21A) - Steroidhormon-Stoffwechsel
Eisen findet sich auch als Cofaktor folgender Enzyme:
- Inositol-Oxygenase - Inositol-Abbau
- Alkohol-Dehydrogenase
- Superoxiddismutase (SOD) - Entgiftung von Superoxiden
- Linoleoyl-CoA-Desaturase - Bildung von Arachidonsäure aus Linolsäure
- Delta(5)-Acyl-CoA-Desaturase - Bildung von Arachidonsäure aus Linolsäure
- Arachidonat-5-Lipoxygenase - Arachidonsäure-Stoffwechsel
- Ribonucleosid-diphosphat- Reduktase - Pyrimidin-Stoffwechsel
- Trimethyllysin-Dioxygenase - Carnitin-Bildung
- γ-Butyrobetain-Dioxygenase - Carnitin-Bildung
- Cystein-Dioxygenase - Taurin-Synthese
- Phenylalanin-Hydroxylase - Bildung von Tyrosin aus Phenylalanin
- 4-Hydroxyphenylpyruvat-Dioxygenase - Tyrosin- und Phenylalanin-Abbau
- Homogentisat-1,2-Dioxygenase - Tyrosin-Abbau
- Tyrosin-Hydroxylase - Katecholamin-Biosynthese
- Tryptophan-Hydroxylase - Serotonin- und Melatonin-Synthese
- 3-Hydroxyanthranilat-3,4-Dioxygenase - Tryptophan-Abbau
- Beta-Caroten-15,15'-Monooxygenase - Retinol-Stoffwechsel
Eisenstoffwechsel
BearbeitenIm Körper befinden sich etwa 3 bis 5 g Eisen, etwa 2/3 davon im Hämoglobin.
Eisen wird im Darm von einem Transporter für divalente Kationen aufgenommen. Ascorbinsäure fördert die Bildung von Fe2+ aus Fe3+ und damit die Eisenresorption. Die Resorptionsquote beträgt etwa 10 % und kann bei Eisenmangel erhöht werden. Der Tagesbedarf beträgt etwa 3 mg bei Männern und 5 mg bei Frauen (Regelblutung, Schwangerschaft und Stillzeit erhöhen den Bedarf).
Im Blutplasma wird Eisen an Transferrin gebunden transportiert („Transporteisen“). Ein Transferrin-Molekül kann 2 Eisenionen aufnehmen.
Gespeichert wird Eisen im Ferritin („Speichereisen“) im retikolendothelialen System (RES) von Leber und Milz und im Knochenmark. Jedes Ferritin-Molekül besteht aus 24 identischen Proteinuntereinheiten, die eine Hohlkugel formen, welche bis über 4.000 Eisenionen speichern kann.
Der zelluläre Eisengehalt wird vom eisensensorischen Protein reguliert.
An der Resorption des Eisens im Dünndarm sind viele weitere Proteine beteiligt, so z.B. Hepcidin, das die Eisenaufnahme hemmt.
Pathobiochemie
Bearbeiten- Eisenmangelanämie - Unzureichenden Häm-Bildung. Labor: S-Transferrin erhöht, S-Transferrin-Sättigung und S-Ferritin vermindert.
- Siderose - Eisenüberladung und Ablagerung im Gewebe in Form des unlöslichen Hämosiderins. Z.B. als Transfusionssiderose.
- Hämochromatose - Siderose durch Gendefekte, z.B. des HFE-Gens (OMIM)
Weblinks
Bearbeiten
Iodid
BearbeitenVorkommen
Bearbeiten| Iodid | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
 | ||||||||||
| Basisdaten und Verweise | ||||||||||
|
Bestandteil der Schilddrüsenhormone Triiodthyronin (T3) und L-Thyroxin (T4).
Pathobiochemie
Bearbeiten- Iodmangel-Struma (endemische Struma)
Zink
BearbeitenVorkommen
Bearbeiten| Zink | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
 | ||||||||||
| Basisdaten und Verweise | ||||||||||
|
- Enzyme (Beispiele):
- Fructose-1,6-bisphosphat-Aldolase
- Pyruvat-Carboxylase
- Alkoholdehydrogenase (NAD+) /(NADP+)
- Cu,Zn-Superoxiddismutase - Entgiftet Superoxide
- Carboanhydrase
- Porphobilinogen-Synthase - Porphyrinbiosynthese
- LTA4-Hydrolase - Biosynthese von LTB4
- Methioninsynthase und Betain--Homocystein-S-Methyltransferase - Rückgewinnung von Methionin aus Homocystein
- L-Dopachrom-Isomerase - Melanin-Bildung
- Membran-Alanyl-Aminopeptidase - γ-Glutamyl-Zyklus
- Alkalische Phosphatase (AP)
- Carboxypeptidase - Pankreasenzym, katalysiert die hydrolytische Spaltung von Peptidbindungen
- DNA-bindende Proteine mit Zinkfinger-, Zinkcluster- oder Zinkdrehungsmotiven. Bsp.: Steroidhormonrezeptoren.
- Speicherform des Insulins (Zink-Insulin-Komplexe)
Pathobiochemie
Bearbeiten- Zink-Mangel
- Acrodermatitis enteropathica (OMIM)
Kupfer
BearbeitenVorkommen
Bearbeiten| Kupfer | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
 | ||||||||||
| Basisdaten und Verweise | ||||||||||
|
- Cytochrom-c-Oxidase - Atmungskette (Komplex IV)
- Kupfer,Zink-Superoxiddismutase (Cu,Zn-SOD) - siehe unter Atmungskette
- Tyrosinase - Tyrosin-Stoffwechsel
- Dopamin-β-Hydroxylase - Tyrosin-Stoffwechsel
- Diamino-Oxidase (DAO) - Tyrosin-, Phenylalanin-, Tryptophan- und Histidin-Stoffwechsel.
Pathobiochemie
Bearbeiten- Frühkindliche Leberzirrhose bei nicht-gestillten Kindern und hohem Kupfergehalt des Trinkwassers (Kupferleitungen, bes. bei saurem Wasser).
- Morbus Wilson (Kupferspeicherkrankheit, Bronzediabetes, hepatolentikuläre Degeneration) - Leberzirrhose, Diabetes mellitus, Kardiomyopathie, neurologische Störungen.
Selen
BearbeitenVorkommen
Bearbeiten| Selen | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
 | ||||||||||
| Basisdaten und Verweise | ||||||||||
|
- Glutathion-Peroxidase - Wasserstoffsuperoxid-Elimination
Pathobiochemie
Bearbeiten- Keshan-Krankheit (Selen-Mangel)
Literatur
BearbeitenPMID 476008 PMID 3920895 PMID 1527993 PMID 3117996 PMID 8793816 PMID 8846151 PMID 3918205 PMID 6401914
Mangan
BearbeitenVorkommen
Bearbeiten| Mangan | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
 | ||||||||||
| Basisdaten und Verweise | ||||||||||
|
- Pyruvat-Carboxylase
- Isocitrat-Dehydrogenase - Citratzyklus
- Superoxiddismutase (SOD) - Entgiftung von Superoxiden
- Katalase - siehe unter Atmungskette
- Phosphoethanolamin-/Phosphocholin-Phosphatase - Bildung von CDP-Cholin
- 3-Isopentenylpyrophosphat-Isomerase - Cholesterinbiosyntheseweg
- Squalen-Synthase - Cholesterinbiosyntheseweg
- Arginase - Harnstoffzyklus
- Galactosyltransferase I - Proteoglykan-Synthese
- Galactosyltransferase II - Proteoglykan-Synthese
- Glucuronosyltransferase I - Proteoglykan-Synthese
Kobalt
BearbeitenVorkommen
Bearbeiten| Kobalt | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
 | ||||||||||
| Basisdaten und Verweise | ||||||||||
|
- Cobalamin (Vitamin B12)
- Phosphoethanolamin-/Phosphocholin-Phosphatase - Bildung von CDP-Cholin
Molybdän
BearbeitenMolybdän wird als Molybdat aufgenommen und ausschließlich bei der Biosynthese der zwei Molybdän-Kofaktoren in Molybdopterin eingebaut.
Vorkommen
Bearbeiten| Molybdän | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
 | ||||||||||
| Basisdaten und Verweise | ||||||||||
|
- Aldehyd-Oxidase - Serotonin-Abbauweg
- Xanthin-Oxidase (XOD) - Purin-Abbau
- Sulfit-Oxidase - Bildung von PAPS
Literatur
Bearbeiten
Magnesium
BearbeitenAllgemeines
BearbeitenMagnesium ist Cofaktor verschiedener Enzyme. Am Muskel wirkt es als physiologischer Kalziumantagonist.
Vorkommen
Bearbeiten| Magnesium | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
 | ||||||||||
| Basisdaten und Verweise | ||||||||||
|
- Enolase
- Phosphatidylinositol-3,4,5-trisphosphat-3-Phosphatase - Inositolphosphat-Stoffwechsel
- Isocitrat-Dehydrogenase - Citratzyklus
- α-Galactosidase - Sphingolipid-Stoffwechsel
- 3-Isopentenyl-pyrophosphat-Isomerase - Cholesterinbiosyntheseweg
- Squalen-Synthase - Cholesterinbiosyntheseweg
- Cholat--CoA-Ligase - Gallensäuren-Stoffwechsel
- Phosphoethanolamin-/Phosphocholin-Phosphatase - Bildung von CDP-Cholin
- 6-Pyruvoyltetrahydropterin-Synthase - Biopterin-Biosynthese
Kalzium
Bearbeiten| Kalzium | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
 | ||||||||||
| Basisdaten und Verweise | ||||||||||
|
Allgemeines
BearbeitenKalzium dient als Bausubstanz von Knochen und Zähnen, als intrazellulärer Botenstoff, als Coaktivator der Blutgerinnung und als Cofaktor verschiedener Enzyme.
Vorkommen und biologische Funktionen
Bearbeiten- Bausubstanz der Knochen und Zähne in Form von Calciumhydroxylapatit ([Ca5OH(PO4)3])
- Second messenger - z.B. bei der Muskelkontraktion oder in anderen Zellen im Rahmen der IP3-Kaskade
- Coaktivator der Blutgerinnung
- Als Cofaktor von Enzymen:
- NAD(P)H-Oxidase - ROS-Bildung
- Apyrase - Pyrimidin- und Purin-Stoffwechsel
- Phospholipase A2 - Phosphoglycerid- und Arachidonsäure-Stoffwechsel
- Inositol-trisphosphat-3-Kinase - Inositolphosphat-Stoffwechsel
- 3-Isopentenyl-pyrophosphat-Isomerase - Cholesterinbiosynthese
Kalzium-Stoffwechsel
BearbeitenIm Körper befinden sich etwa 1 kg Kalzium, 99 % davon im Knochen. Das Skelett dient als inneres Stützsystem des Körpers, aber auch als Kalziumspeicher, um den Blutkalziumspiegel konstant zu halten. Der Tagesbedarf eines Erwachsenen beträgt etwa 1 g. Der Kalzium-Stoffwechsel wird von den folgenden Hormonen kontrolliert:
Anhebung der S-Kalzium-Spiegels durch
- Parathormon (PTH, aus der Nebenschilddrüse)
- -> Aktiviert (indirekt) die Osteoklasten und hemmt die Osteoblasten.
- -> Fördert in der Niere die Calcium- und hemmt die Phosphat-Rückresorption.
- -> Fördert die Calcitriol-Bildung in der Niere.
- Prolactin (aus der Adenohypophyse)
- -> Fördert die Calcitriol-Bildung in der Niere.
- Vitamin D-Hormon (Calcitriol, 1,25-Dihydroxycholecalciferol, 1,25-(OH)2-D3)
- -> Fördert direkt den Knochenabbau.
- -> Fördert die intestinale Kalzium-Aufnahme, geringer auch die Phosphat-Aufnahme. Dadurch indirekt Förderung des Knochenaufbaus.
- -> Fördert die Kalzium- und Phosphat-Rückresorption in der Niere. Dadurch indirekt Förderung des Knochenaufbaus.
Senkung des S-Kalzium-Spiegels durch
- Calcitonin (aus den C-Zellen der Schilddrüse) - Gegenspieler des Parathormons
- -> Hemmt die Osteoklasten.
- -> Hemmt die Kalzium-Aufnahme im Darm.
- -> Hemmt die Kalzium- und Phosphat-Aufnahme in der Niere.
Pathiobiochemie
BearbeitenHypokalzämie
- Hypoparathyreoidismus z.B. bei Strumektomie, wenn unbeabsichtigt alle Nebenschilddrüsen mitentfernt wurden.
- Kalziummangel (Vitamin-D-Mangel)
Klinik: Hypokalzämische Tetanie durch erhöhte neuromuskuläre Erregbarkeit - Parästhesien und Pfötchenstellung der Hände, Chvostek-Zeichen (Beklopfen des Nervus facialis vor dem Ohr -> Zuckungen im Gesicht).
Hyperventilationstetanie
- Hyperventilation (z.B. bei Aufregung) -> Vermehrte Abatmung von CO2 -> metabolische Alkalose -> H+-Ionen lösen sich von den negativ geladenen Gruppen (z.B. Carboxylgruppen) der Serumproteine und des Hämoglobin ab -> Ca2+-Ionen lagern sich stattdessen dort an -> Abnahme des ionisierten Anteils des S-Kalziums -> Funktionelle Hypokalzämie.
Klinik: wie oben.
Knochenerkrankungen:
- Verminderung der Knochenmineralisierung bei normaler oder vermehrter Interzellularsubstanz
- Rachitis - Kindlicher Vitamin-D-Mangel/Lichtmangel
- Osteomalazie - Erwachsener Vitamin-D-Mangel/Lichtmangel, Kalziummangel
- Verminderung der Knochenmineralisierung und der Interzellularsubstanz
- Osteopenie, Osteoporose - Vitamin-D-Mangel/Lichtmangel, Kalziummangel, Alter, hormonelle Faktoren (Glucokortikoide fördern den Knochenabbau, Östrogene fördern den Knochenaufbau)
Hyperkalziämie bei:
- Hyperparathyreoidismus (HPT)
- Malignome (Osteolysen, Sekretion von parathormone-related peptide)
- Immobilisation (Knochenresorption)
- Sarkoidose (Calcitriol-Bildung durch aktivierte Makrophagen)
- Familiäre hyperkalziurische Hyperkalziämie (OMIM)
- Renal-tubuläre Azidose Typ I (OMIM)
Klinik: „Stein-, Bein- und Magen-Pein“ (s.u.)
Hyperparathyreoidismus (HPT, Überfunktion der Nebenschilddrüse):
- Primärer HPT: NSD-Adenome, NSD-Karzinom -> PTH-Anstieg -> Hyperkalziämie, Hypophosphatämie, erhöhte alkalische Phosphatase.
- Sekundärer HPT: Chron. Niereninsuffizienz, Malabsorption -> Calcitriol-Mangel -> Kalzium-Defizit -> PTH-Anstieg, NSD-Hyperplasie -> Hypo- oder Normokalziämie, Hyper- oder Normophosphatämie, alkalische Phosphatase im Serum erhöht.
- Tertiärer HPT: Fixierter sekundärer HPT durch zunehmende Autonomie des hyperplastischen NSD-Gewebes -> Hyperkalziämie, Hypophosphatämie, erhöhte alkalische Phosphatase.
- Pseudohyperparathyreoidismus: Sekretion von parathormone-related peptide durch Malignome -> Hyperkalziämie, Hypophosphatämie, erhöhte alkalische Phosphatase.
Klinik: Beim prim. und tert. HPT „Stein-, Bein- und Magen-Pein“: Hyperkalzämie, Osteoporose, Akroosteolysen, Knochenschmerzen, Polyurie, Nierensteine (Hyperkalziurie), Weichteilverkalkungen, Gastritis, Pankreatitis, Obstipation, psychiatrische Symptome (Adynamie, Depression), HRST. Beim sek. HPT dominieren die Symptome der Grunderkrankung.
Schwefel
BearbeitenAllgemeines
Bearbeiten| Schwefel | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
 | ||||||||||
| Basisdaten und Verweise | ||||||||||
|
Schwefel ist ein Bestandteil der Aminosäuren Methionin und Cystein und der Cofaktoren Thiamin (Vitamin B1), Biotin, Liponsäure und Molybdän-Cofaktor.
Eisen-Schwefel-Zentren finden sich in folgenden Enzymen:
- Aconitase - Citratzyklus
- Eisensensorisches Protein
- NADH-Dehydrogenase - Atmungskette (Komplex I)
- Succinat-Dehydrogenase - Atmungskette (Komplex II)/Citratzyklus
- Cytochrom-c-Reduktase - Atmungskette (Komplex III)
- Xanthin-Oxidase (XOD) - Harnsäure-Abbau
In Form von Sulfat wird Schwefel insbesondere in Proteoglycane (Glycosaminoglycane) eingebaut. Sulfat wird außerdem in der Leber an lipophile Substanzen geheftet, die ausgeschieden werden sollen (Biotransformation Phase II).
Bindung und Weitergabe von Sulfat
Bearbeiten| ⇓ | Subst. | ( ⇑ ) | Co. | Enzym | EC | EG | Erkr. | |||
|---|---|---|---|---|---|---|---|---|---|---|
|
||||||||||
| O2, H2O
H2O2 |
Häm, Mo | Sulfit-Oxidase | 1.8.3.1 | Ox | Sulfocysteinurie | |||||
|
||||||||||
| ATP
PPi |
Sulfat-Adenylyltransferase | 2.7.7.4 | Tr | Spondyloepimetaphyseal dysplasia, Pakistani type (SEMD) | ||||||
|
||||||||||
| Pi
H2O |
PAPS-3'(2')- Phosphohydrolase | 3.1.3.7 | Hyd | |||||||
|
ADP |
oder
|
ADP |
oder
APS-Kinase |
2.7.1.25 | Tr | Spondyloepimetaphyseal dysplasia, Pakistani type (SEMD) | ||||
|
||||||||||
| Phenol
Aryl-Sulfat |
Aryl-Sulfotransferase | 2.8.2.1 | Tr | |||||||
|
Alkyl-Sulfat |
oder
|
oder
Alkohol-Sulfotransferase |
2.8.2.2 | Tr | ||||||
|
Estron-3-Sulfat |
oder
|
oder
Estron-Sulfotransferase |
2.8.2.4 | Tr | ||||||
|
Chondroitin-4-sulfat |
oder
|
oder
Chondroitin- 4-Sulfotransferase |
2.8.2.5 | Tr | ||||||
|
Sulfat |
oder
|
oder
Nukleotid-Diphosphatase |
3.6.1.9 | Hyd | IIAC, ARHR2, OPLL | |||||
|
||||||||||




{kind=link}
PAPS dient als Sulfatgruppen-Lieferant
BearbeitenPAPS wird als „aktiviertes Sulfat“ für Sulfatierungen benötigt, z.B. im Keratansulfat-, Chondroitinsulfat- und im Heparansulfat-Stoffwechsel und in der Biotransformation Phase II (Sulfatierung von OH-Gruppen).
Literatur
BearbeitenWeblinks
Bearbeiten
Biotransformation
BearbeitenAllgemeines
BearbeitenDie Biotransformation von körpereigenen und körperfremden Stoffen findet bevorzugt im glatten endoplasmatischen Retikulum der Darmmucosa- und Leberzellen statt. Dabei werden vor allem lipophile Substanzen umgesetzt. Ziel ist die Erhöhung der Wasserlöslichkeit, so dass die Stoffe leichter über die Galle oder den Urin ausgeschieden werden können. Das System dient auch der Bildung wichtiger Metabolite, wie z.B. verschiedener Steroidhormone und Vitamin D-Hormon.
Phase I (Umwandlungsreaktionen)
BearbeitenEinfügung oder Veränderung funktioneller Gruppen.
Hydrolytische Spaltung
BearbeitenMethylierung
BearbeitenRedoxreaktionen
BearbeitenAm wichtigsten sind Oxidationsvorgänge (z.B. Hydroxylierungen), die v.a. von Cytochrom-P450-abhängigen Enzymen (CYP) realisiert werden. Das Häm-Thiolat-haltige Cytochrom-P450 dient dabei den Monooxygenasen (Hydroxylasen) als Elektronenüberträger.
Enzyme (Beispiele):
- CYP1A1
- Unspezifische Monooxygenase (EC 1.14.14.1) - Steroidhormon-Stoffwechsel
- CYP1A2
- Unspezifische Monooxygenase (EC 1.14.14.1) - Steroidhormon-Stoffwechsel
- CYP1B1
- Unspezifische Monooxygenase (EC 1.14.14.1) - Steroidhormon-Stoffwechsel
- CYP2A
- Unspezifische Monooxygenase (EC 1.14.14.1) - Steroidhormon-Stoffwechsel
- CYP2B
- Unspezifische Monooxygenase (EC 1.14.14.1) - Steroidhormon-Stoffwechsel
- CYP2C
- Unspezifische Monooxygenase (EC 1.14.14.1) - Steroidhormon-Stoffwechsel
- CYP2D
- Unspezifische Monooxygenase (EC 1.14.14.1) - Steroidhormon-Stoffwechsel
- CYP2E
- Unspezifische Monooxygenase (EC 1.14.14.1) - Steroidhormon-Stoffwechsel
- CYP2F
- Unspezifische Monooxygenase (EC 1.14.14.1) - Steroidhormon-Stoffwechsel
- CYP2J
- Unspezifische Monooxygenase (EC 1.14.14.1) - Steroidhormon-Stoffwechsel
- CYP2S
- Unspezifische Monooxygenase (EC 1.14.14.1) - Steroidhormon-Stoffwechsel
- CYP3A
- Unspezifische Monooxygenase (EC 1.14.14.1) - Steroidhormon-Stoffwechsel
- CYP4B
- Unspezifische Monooxygenase (EC 1.14.14.1) - Steroidhormon-Stoffwechsel
- CYP4X
- Unspezifische Monooxygenase (EC 1.14.14.1) - Steroidhormon-Stoffwechsel
- CYP4Z
- Unspezifische Monooxygenase (EC 1.14.14.1) - Steroidhormon-Stoffwechsel
- CYP5A
- Thromboxan-A-Synthase (EC 5.3.99.5) - Biosynthese von TXA2
- CYP7A1
- Cholesterol-7α-Monooxygenase (EC 1.14.13.17) - Gallensäuren-Biosynthese
- CYP8A
- Prostacyclin-Synthase (EC 5.3.99.4) - Biosynthese von Prostacyclin
- CYP11A
- Cholesterol-Monooxygenase (20-22-Desmolase) (EC 1.14.15.6) - Steroidhormon-Stoffwechsel
- CYP11B
- Steroid-11β-Monooxygenase (11β-Hydroxylase) (EC 1.14.15.4) - Steroidhormon-Stoffwechsel
- Corticosteron-18-Monooxygenase (18-Hydroxylase) (EC 1.14.15.5) - Steroidhormon-Stoffwechsel
- CYP17A
- Steroid-17α-Monooxygenase (17α-Hydroxylase) (EC 1.14.99.9) - Steroidhormon-Stoffwechsel
- CYP19A
- Unspezifische Monooxygenase (EC 1.14.14.1) - Steroidhormon-Stoffwechsel
- CYP21A
- Steroid-21-Monooxygenase (21-Hydroxylase) (EC 1.14.99.10) - Steroidhormon-Stoffwechsel
- CYP24A
- 25-Hydroxyvitamin-D3-24-Hydroxylase
- CYP27A
- Cholestanetriol-26-Monooxygenase (EC 1.14.13.15) - Gallensäuren-Biosynthese
- CYP27B
- Calcidiol-1-Monooxygenase (EC 1.14.13.13) - Calcitriol-Bildung
- CYP51A
- Sterol-14-Demethylase (EC 1.14.13.70) - Cholesterin-Biosynthese
Phase II (Konjugationsreaktionen)
BearbeitenKopplung an hydrophile Moleküle.
Glucuronidierung
BearbeitenKonjugation mit Glucuronat - Glucuronidierung von OH- und NH2-Gruppen mit Hilfe von UDP-D-Glucuronat.
Enzyme:
- Glucuronosyltransferase (EC 2.4.1.17)
Sulfatierung
BearbeitenKonjugation mit Sulfat - Sulfatierung von OH-Gruppen mit Hilfe von 3'-Phosphoadenosin-5'-phosphosulfat (PAPS).
Enzyme:
- Aryl-Sulfotransferase (EC 2.8.2.1)
- Alkohol-Sulfotransferase (EC 2.8.2.2)
- Estron-Sulfotransferase (EC 2.8.2.4)
Acetylierung
BearbeitenKonjugation mit Acetat - Acetylierung von NH2-Gruppen.
Amidierung
BearbeitenKonjugation mit Aminosäuren.
Glutathionylierung
BearbeitenKonjugation mit Glutathion und Bildung von Mercapturaten.
- Metabbolite - z.B. Östrogene, Melanin, Prostaglandine, Leukotriene
- Xenobiotika - z.B. Bromobenzen, Acetaminophen
Enzyme:
- Glutathion-S-Transferase (2.5.1.18)
Pathobiochemie
Bearbeiten- Giftung - Manche an sich u.U. harmlose Fremdstoffe werden erst durch die Biotransformation zu giftigen z.T. krebserregenden Substanzen umgesetzt.
- Lebererkrankungen - Bei Lebererkrankungen wie der Leberzirrhose kann es durch Reduktion der Biotransformationskapazität zu hormonellen Störungen kommen, z.B. zur Gynäkomastie durch Steroidhormon-Akkumulation und verstärkte Umwandlung von Androgenen zu Östrogenen im Fettgewebe.
Pharmakologie
BearbeitenViele (bes. lipophile) Arzneistoffe werden nach der Resorption aus dem Verdauungstrakt bereits bei der ersten Leberpassage mehr oder weniger stark abgebaut, so dass ein Teil nicht mehr am Zielort ankommen kann. Dieser First-Pass-Effekt muss bei der Dosierung berücksichtigt werden.
Die Induktion und Hemmung der Enzyme oder die Konkurrenz am Enzym ist für zahlreiche pharmakologische Wechselwirkungen zwischen Medikamenten, manchen Nahrungsmitteln (Grapefruitsaft) und Alkohol verantwortlich. Die individuelle Enzymausstattung jedes Menschen kann weiterhin zu Dosierungsproblemen führen, da die Medikamente unterschiedlich schnell verstoffwechselt werden.
Weblinks
Bearbeiten- KEGG: Cytochrome P450
- KEGG: Xenobiotics Biodegradation and Metabolism
- Flockhart DA. Drug Interactions: Cytochrome P450 Drug Interaction Table. Indiana University School of Medicine (2007)
- RCSB PDB: Cytochrome p450
- RCSB PDB: Sulfotransferases
Hämoglobin
BearbeitenAllgemeines
Bearbeiten |
 |
 |
 |
Der rote Blutfarbstoff Hämoglobin (Hb) ist für den Sauerstofftransport von der Lunge in die Peripherie verantwortlich. Das Hämoglobin befindet sich in den Erythrozyten (MCH: ca. 27 - 34 pg Hämoglobin pro Erythrozyt) und bildet etwa 1/3 der Masse der Erythrozyten (MCHC: 320 - 360 g/l). Im Vollblut bildet Hämoglobin die Hauptmasse an Protein (Hb: 14 - 18 g/dl, im Vergleich dazu das Serumprotein: 6,5 - 8,5 g/dl).
Aufbau
BearbeitenDas Hämoglobin-Molekül ist ein Tetramer aus 4 Untereinheiten. Jede Untereinheit besteht aus einem Häm und einem Protein-Anteil, der Globinkette. Die Globinkettengene (α, β, γ, δ, ε, ξ) werden abhängig vom Entwicklungsstadium exprimiert. Die in utero vorherrschenden Hämoglobine besitzen eine höhere Sauerstoffaffinität, um den Sauerstofftransfer vom mütterlichen ins kindliche Blut an der Plazentarschranke zu unterstützen.
HB-Typen nach Art der Globin-Kette
Bearbeiten- HbA1 (α2β2) - 97 % des Hb beim Gesunden
- HbA2 (α2δ2) - 2,5 % des Hb beim Gesunden
- HbF (α2γ2) - Fetales Hämoglobin
- Gower 1 (ξ2ε2) - Embryonales Hämoglobin
- Gower 2 (α2ε2) - Embryonales Hämoglobin
- Hämoglobin Portland (ξ2γ2) - Embryonales Hämoglobin
Funktionen
Bearbeiten- Sauerstofftransport. 1 g Hb bindet in vivo maximal 1,34 ml O2 (Hüfner-Zahl). Umgerechnet in mmol heißt das: 1 mmol Tetramer (64,5 g) bindet maximal 86,34 ml O2.
- Säure-Puffer durch Bindung von H+ v.a. im Kapillarbett. Protonen werden bei der Umwandlung von CO2 in HCO3- durch die Carboanhydrase frei. Im Lungengefäßbett erfolgt die Rückreaktion und Abgabe von Kohlendioxid zur Abatmung.
- Bindung von CO2 über Carbaminobindungen und Transport Richtung Lunge (etwa 20 % des CO2). Desoxygeniertes Hb bindet CO2 besser als oxygeniertes Hb.
Kooperativität und Sauerstoffbindungskurve
BearbeitenNormalerweise würde man beim Hämoglobin wie bei vielen Enzymen eine hyperbolische Substrat-Bindungskurve erwarten. Real bietet sich jedoch ein sigmoider Verlauf. Dies erklärt sich aus der Kooperativität der vier Untereinheiten. Bindet das Häm einer Untereinheit ein Molekül Sauerstoff (O2), so kommt es zu einer Konformationsänderung der vier miteinander verbundenen Hb-Untereinheiten, die die Anlagerung von weiteren Sauerstoffmolekülen an die anderen Untereinheiten erleichtert. Dadurch verläuft die O2-Bindungskurve im physiologischen Bereich der Sauerstoffpartialdrücke steiler und das Hämoglobin kann im Lungenkreislauf leichter mit Sauerstoff beladen werden und ihn im peripheren Gewebe leichter abgeben.
Modulation des Sauerstoffbindungskurve
BearbeitenDas Verhalten des Hämoglobins wird noch dadurch optimiert, dass die Sauerstoffbindungskurve von verschiedenen Effektoren beeinflusst werden kann. Zu einer Rechtsverschiebung der Kurve im peripheren Kapillarbett mit Abnahme der O2-Affinität bzw. Erleichterung der O2-Abgabe kommt es durch:
- die Bindung von Protonen und CO2 (bekannt als sog. Bohr-Effekt).
- die Bindung von 2,3-Bisphosphoglycerat. 2,3-BPG entsteht in einem Nebenweg der Glycolyse aus 1,3-BPG. Es bindet allosterisch v.a. an Desoxy-Hb und stabilisiert es dadurch. Fetales HbF bindet 2,3-BPG geringer als adultes Hb, da es eine höhere Sauerstoffaffinität braucht, um in der Plazenta genügend Sauerstoff aus dem mütterlichen Kreislauf abzuziehen.
- steigender Temperatur (z.B. im arbeitenden Muskel)
Eine Linksverschiebung der Kurve in der Lungenstrombahn mit Erhöhung der O2-Affinität kommt dadurch zustande, dass hier die Protonen- und CO2-Konzentration abnimmt.
Strukturell verwandte Proteine
BearbeitenDas Myoglobin dient als Sauerstoffspeicher im Muskel (v.a. in roten Typ-I-Muskelfasern). Es entspricht in seinem Aufbau einer Hämoglobin-Untereinheit und zeigt daher keine Kooperativität. Myoglobin besitzt eine höhere Sauerstoffaffinität als Hämoglobin.
Pathobiochemie und Labormedizin
Bearbeiten- Anämie - Einen Mangel an Hämoglobin bzw. Erythrozyten nennt man Anämie (Blutarmut). Diese kann z.B. durch Blutbildungsstörungen bedingt sein (Eisen-Mangel, Folat-Mangel, Cobalamin-Mangel, Vergiftung mit Benzol oder Blei, erbliche Defekte wie die Thalassämie, Eisenverwertungsstörungen bei SIRS, Knochenmarksverdrängung), durch erhöhte Verluste (Blutung) oder vorzeitigen Abbau (Autoantikörper, Membrandefekte (Kugelzellanämie), Enzymdefekte wie z.B. dem (G6PD-Mangel)).
- Sichelzellanämie (OMIM) - Das Hb ist qualitativ verändert (HbA1 -> HbS). Die Mutation der β-Kette (Ersatz von Glutamat gegen Valin in Position 6) bewirkt eine Polymerisation des Hämoglobins v.a. im venösen Blut und damit ein Aussicheln der Erythrozyten. Die Mutation bewirkt eine höhere Malaria-Resistenz und konnte sich wahrscheinlich deswegen so gut verbreiten.
- Thalassämie - Das Hb ist quantitativ verändert. Die Synthese einer oder mehrerer Hb-Ketten ist gestört. Je nach betroffener Globinkette und Schweregrad kommt es zur ineffektiven Erythropoese (Blutbildung) mit Anämie (Blutarmut), einer vermehrten Bildung fetaler und embryonaler Hämoglobine, Ikterus (Gelbsucht), Gallenstein-Bildung durch vermehrten Bilirubin-Anfall und Knochenveränderungen sowie Leber- und Milzvergrößerung durch die Expansion der Erythropoese. β-Thalassämien (OMIM) führen schneller zu Symptomen als α-Thalassämien (OMIM), da es vier α-Kettengene, aber nur zwei β-Kettengene gibt.
- HbA1c - Die nicht-enzymatische Glycosylierung des Hämoglobins korreliert mit dem Blutzuckerspiegel. Aufgrund der langen Erythrozyten-Lebensdauer von ca. 120 Tagen kann der HbA1c-Wert als „Blutzuckergedächtnis“ angesehen und als Indikator für die Güte der mittelfristigen Blutzuckereinstellung beim Diabetiker genutzt werden.
Weblinks
Bearbeiten
Übungsfragen
BearbeitenIm Folgenden finden Sie einige Übungs- und Wiederholungsfragen. Wenn man davon viele nicht auf Anhieb beantworten kann, dann sollte man sich davon nicht beunruhigen lassen. Übung macht den Meister und zum Üben sind die Fragen da!
Fragen 1 - 10
BearbeitenWelche zwei Stoffwechselwege bilden die zentrale Achse des Intermediärstoffwechsels?
- Glycolyse/Gluconeogenese
- Citratzyklus
Wie heißt das stickstoffhaltige Endprodukt des Aminosäuren-Stoffwechsels?
Harnstoff
Wie heißt das stickstoffhaltige Endprodukt des Purin-Stoffwechsels?
Harnsäure
Wie heißt der „Universalbrennstoff“ des Zitronensäurezyklus?
Acetyl-CoA (aktivierte Essigsäure)
Mit welchem Intermediat des Citratzyklus und welcher Aminosäure beginnt die Biosynthese von Häm?
Succinyl-CoA und Glycin
Welche Stoffe kann der Körper aus Cholesterin bilden? Nennen Sie zwei Beispiele!
- Hormone, z.B. Kortisol, Aldosteron, Testosteron, Östrogen, Progesteron
- Gallensäuren wie Taurocholat und Glycocholat
(Hinweis: Vitamin D-Hormon wird aus dem Cholesterinvorläufer 7-Dehydrocholesterol (Provitamin D3) gebildet.)
Wie heißt das Abbauprodukt des Häms, das bei Leberschädigung, Gallenstau oder Hämolyse zur typischen Gelbfärbung (Ikterus) von Haut und Skleren sowie zum typischen bierbraunen Urin führt?
Bilirubin
Welche Aminosäure entsteht durch Transaminierung aus Pyruvat?
Alanin. Das verantwortliche Enzym ist die Alanin-Transaminase (ALT, ALAT, GPT).
Welche wichtigen Substanzen entstammen dem Tyrosin-Stoffwechsel?
- Die Schilddrüsenhormone Triiodthyronin (T3) und L-Thyroxin (T4)
- Die Katecholamine Dopamin, Noradrenalin und Adrenalin
- Das Hautpigment Melanin
Wozu dient der Hexosemonophosphatweg (Pentosephosphat-Shunt)? Nennen Sie zwei wichtige Funktionen!
- Generierung von NADPH/H+
- Erzeugung von D-Ribose-5-phosphat für die Purin- und Pyrimidin-Biosynthese.
Fragen 11 - 20
BearbeitenWelche zwei Botenstoffe können aus der Aminosäure Tryptophan gebildet werden?
Serotonin und Melatonin
Welche Funktion hat Glucuronsäure?
Erhöhung der Wasserlöslichkeit von körpereigenen oder körperfremden Stoffen, die ausgeschieden werden sollen. Diese Konjugationsvorgänge (Biotransformation Phase II) finden vor allem in der Leber statt.
Welche Aminosäure entsteht durch Transaminierung aus Oxalacetat?
Aspartat. Das verantwortliche Enzym ist die Aspartat-Transaminase (AST, ASAT, GOT).
Wie wird Ethanol abgebaut?
Ethanol -> Acetaldehyd -> Acetat -> Acetyl-CoA -> Einschleusung z.B. in Citratzyklus oder Fettsäurenbiosynthese
Nennen Sie 3 biogene Amine, die durch PALP-abhängige Decarboxylierung aus einer proteinogenen Aminosäure oder aus einem Derivat einer proteinogenen Aminosäure gebildet werden!
Direkt aus proteinogenen Aminosäuren gehen hervor:
- γ-Aminobuttersäure (GABA) aus Glutamat
- Histamin aus Histidin
Aus Derivaten proteinogener Aminosäuren gehen hervor:
- Tyramin aus Tyrosin
- Dopamin aus L-Dopa
- Tryptamin aus L-Tryptophan
- Serotonin (5-Hydroxy-Tryptamin) aus 5-Hydroxy-Tryptophan
Wie heißt der wichtigste allosterische Regulator von Glycolyse und Glucogenese?
Fructose-2,6-bisphosphat. Das Molekül aktiviert die Phosphofructokinase (Glycolyse) und hemmt die Fructose-1,6-bisphosphatase (Gluconeogenese).
Nennen Sie zwei Stoffwechselwege, die durch Glucagon aktiviert werden!
- Gluconeogenese
- Glycogenolyse
Wofür benötigt der Körper Folsäure?
Synthese von DNA- und RNA-Bausteinen:
- Synthese von Purinen
- Synthese von Thymidylat
Welcher Stoffwechselschritt benötigt sowohl Folsäure als auch Cobalamin?
Die Remethylierung von Homocystein zu Methionin.
Nennen Sie zwei Stoffwechselwege die überwiegend im Mitochondrium lokalisiert sind!
- Zitratzyklus
- β-Oxidation der Fettsäuren
Fragen 21 - 30
BearbeitenWie heißt das Schrittmacherenzym der Cholesterinbiosynthese? Durch welche Medikamentengruppe kann es inhibiert werden?
- Das geschwindigkeitsbestimmende Enzym ist die HMG-CoA-Reduktase.
- Hemmstoffe heißen HMG-CoA-Reduktase-Hemmer / CSE-Hemmer / Statine.
In welchem Abbauweg entsteht Vanilinmandelsäure?
Katecholamin-Abbau
In welchem Abbauweg entsteht Hydroxyindolessigsäure?
Serotonin-Abbau
In welchem Stoffwechselweg entsteht Kohlenmonoxid?
Abbau von Häm zu Biliverdin
Aus welcher Fettsäure werden Prostaglandine und Leukotriene gebildet?
Arachidonsäure, eine 4fach ungesättigte ω-6-Fettsäure
Wie heißt das biologisch aktive Vitamin D, das durch die zweimalige Hydroxylierung von Vitamin D3 in Leber und Niere entsteht?
1,25-Dihydroxycholecalciferol (Calcitriol)
Welches Vitamin spielt als Coenzym der Dehydrogenase-Komplexe und der Transketolase eine wichtige Rolle?
Thiamin bzw. Thiamindiphosphat (Vitamin B1).
Welches Vitamin ist typischerweise an Carboxylierungsreaktionen beteiligt?
Biotin (Vitamin H)
Pantothensäure (Vitamin B5) ist Bestandteil welches wichtigen Trägermoleküls?
Pantothensäure ist ein Bestandteil von Coenzym A
Ordnen Sie die Enzyme der 1. Spalte den entsprechenden Stoffgruppen in der 2. Spalte zu!
| Enzym | Stoffgruppe |
| a) Cyclooxygenase | 1) Eikosanoide |
| b) Phospholipase A2 | 2) Leukotriene |
| c) Lipoxygenase | 3) Prostaglandine |
a-3, b-1, c-2
Die Phospholipase A2 spaltet Arachidonsäure von Membran-Phospholipiden ab und steht damit am Anfang der Eikosanoid-Synthese. Die Cyclooxygenase (COX) wird danach für die Bildung der Prostaglandine benötigt, Lipoxygenasen katalysieren die Synthese der Leukotriene.
Fragen 31 - 40
BearbeitenWie heißen die drei verzweigtkettigen Aminosäuren?
Valin, Leucin und Isoleucin
Welches Molekül ist für den Transport von Fettsäuren ins Mitochondrium zuständig? Aus welcher Aminosäure wird es gebildet?
Fettsäuren werden mit Carnitin verestert, damit sie die innere Mitochondrienmembran überwinden können. Gebildet wird Carnitin aus (proteingebundenem) Lysin.
Welches Molekül, das insbesondere im Skelettmuskel als temporärer Energiespeicher benötigt wird, wird aus Arginin und Glycin gebildet?
Kreatinphosphat
Aspartat liefert Stickstoff unter Abspaltung von Fumarat in welche zwei Stoffwechselwege (Aspartatzyklus)?
- Purinbiosynthese (Bildung von IMP und von AMP aus IMP)
- Harnstoffzyklus
Ordnen Sie die Enzyme der 1. Spalte den spezifischen Hemmstoffen der 2. Spalte zu!
| Enzym | Hemmstoff |
| a) Cyclooxygenase (COX) | 1) Leflunomid |
| b) Hydroxymethylglutaryl-CoA-Reduktase (HMG-CoA-Reduktase) | 2) Finasterid |
| c) Dihydroorotat-Dehydrogenase | 3) Simvastatin |
| d) Thymidylatkinase | 4) Acetylsalicylsäure (ASS) |
| e) 5α-Reduktase | 5) 5-Fluoruracil (5-FU) |
Richtige Zuordnung:
a) Cyclooxygenase (Prostaglandinsynthese) - 4) Acetylsalicylsäure (ASS)
b) Hydroxymethylglutaryl-CoA-Reduktase (Cholesterinsynthese) - 3) Simvastatin
c) Dihydroorotat-Dehydrogenase (Pyrimidin-Synthese) - 1) Leflunomid
d) Thymidylatkinase (Thymidin-Synthese) - 5) 5-Fluoruracil (5-FU)
e) 5α-Reduktase (Dihydrotestosteron-Bildung) - 2) Finasterid
Ordnen Sie die Enzyme der 1. Spalte den spezifischen Hemmstoffen der 2. Spalte zu!
| Enzym | Hemmstoff |
| a) Xanthin-Oxidase (XOD) | 1) Warfarin |
| b) Aromatase | 2) Allopurinol |
| c) Vitamin-K-Epoxid-Reduktase | 3) Parathion (E605) |
| d) Monoamino-Oxidase A (MAO A) | 4) Methotrexat (MTX) |
| e) Acetylcholinesterase | 5) Moclobemid |
| f) Dihydrofolat-Reduktase (DHFR) | 6) Anastrozol |
Richtige Zuordnung:
a) Xanthinoxidase (Harnsäuresynthese) - 2) Allopurinol
b) Aromatase (Östrogen-Synthese) - 6) Anastrozol
c) Vitamin-K-Epoxid-Reduktase (γ-Carboxylierung der Gerinnungsfaktorvorstufen) - 1) Warfarin
d) Monoamino-Oxidase A (Katecholamin- und Serotonin-Abbau) - 5) Moclobemid
e) Acetylcholinesterase (Acetylcholin-Abbau) - 3) Parathion
f) Dihydrofolat-Reduktase (Folat-Stoffwechsel) - 4) Methotrexat (MTX)
Ordnen Sie die Enzyme der 1. Spalte den spezifischen Hemmstoffen der 2. Spalte zu!
| Enzym | Hemmstoff |
| a) Catechol-O-Methyltransferase (COMT) | 1) Acarbose |
| b) α-Glucosidase | 2) Blausäure |
| c) DOPA-Decarboxylase | 3) Acetazolamid |
| d) Cytochrom-c-Oxidase | 4) Entacapon |
| e) Carboanhydrase (CA) | 5) Carbidopa, Benserazid |
Richtige Zuordnung:
a) Catechol-O-Methyltransferase (COMT) - 4) Entacapon
b) α-Glucosidase - 1) Acarbose
c) DOPA-Decarboxylase - 5) Carbidopa, Benserazid
d) Cytochrom-c-Oxidase - 2) Blausäure
e) Carboanhydrase (CA) - 3) Acetazolamid
In welchem Biomolekül findet sich Kobalt?
Cobalamin (Vitamin B12)
Welches eher ungewöhnliche Element findet sich in der Glutathion-Peroxidase?
Das Halbmetall Selen
Welche drei Möglichkeiten gibt es im Stoffwechsel, um Moleküle mit einer zusätzlichen Ein-Kohlenstoff-Gruppe auszustatten?
- Carboxylgruppen können durch Biotin-abhängige Carboxylierung angeheftet werden
- S-Adenosyl-methionin überträgt Methylgruppen
- Folsäure überträgt Formyl- und Methylgruppen
Fragen 41 - 50
BearbeitenDurch die Kopplung an welche Stoffwechselwege wird der C1-Rest der Tetrahydrofolsäure regeneriert?
- Glycin- und Serin-Abbau
- Histidin-Abbau (FIGLU zu Glutamat)
- Formiat-Bindung
Wofür wird Thioredoxin benötigt?
Für die Bildung von Desoxyribonukleotiden.
Welche Faktoren bewirken eine Rechtsverschiebung der Sauerstoffbindungskurve des Hämoglobins? (4)
- Protonen (sinkender pH)
- Kohlendioxid
- Wärme
- 2,3-BPG
Wie lässt sich eine sigmoide Substrat-Bindungskurve am besten erklären?
Durch eine Kooperativität mehrerer Proteinuntereinheiten.
In der Phase II der Biotransormation können Xenobiotika mit welchen Molekülen konjugiert werden? (4)
Glucuronat, Sulfat, Acetat, Aminosäuren
L-Aminosäuren gehen in die Biosynthese verschiedener Moleküle ein. Ordnen Sie den Aminosäuren der 1. Spalte die (am ehesten zutreffenden) Produkte der 2. Spalte zu!
| Aminosäure | Produkt |
| a) Glycin | 1) Carnitin |
| b) Lysin | 2) Taurin, Coenzym A, Glutathion |
| c) Serin | 3) Häm |
| d) Arginin und Glycin | 4) Sphingolipide |
| e) Cystein | 5) Kreatin |
Richtige Zuordnung:
a) Glycin - 3) Häm
b) Lysin - 1) Carnitin
c) Serin - 4) Sphingolipide
d) Arginin und Glycin - 5) Kreatin
e) Cystein - 2) Taurin, Coenzym A, Glutathion
Wie kommen Reduktionsäquivalente (NADH/H+) vom Zytosol ins Mitochondrium? (2)
Mit dem Malat-Aspartat-Shuttle oder mit dem Glycerin-3-Phosphat-Shuttle.
Welches FAD-haltige Enzym des Krebs-Zyklus ist ein Bestandteil der mitochondrialen Atmungskette?
Die Succinat-Dehydrogenase (Komplex II der Atmungskette)
Aus welcher Aminosäure wird von der NO-Synthase Stickoxid (NO, EDRF) freigesetzt?
Aus Arginin
Was haben cAMP, cGMP, Diacylglycerin (DAG), Inositoltrisphosphat (IP3) und Kalzium gemeinsam ?
Es handelt sich um intrazelluläre Botenstoffe.
- cAMP wird von der Adenylatcyclase gebildet.
- cGMP wird von der Guanylatcyclase gebildet.
- DAG und IP3 werden von der Phospholipase C aus Phosphatidylinositolbisphosphat (PIP2) freigesetzt.
- Kalzium wird aus dem endoplasmatischen Retikulum freigesetzt (Skelettmuskel) oder strömt durch Ionenkanäle aus dem Extrazellularraum ein (Herzmuskel).
Fragen 51 - 60
BearbeitenBringen Sie die folgenden Stoffe/Metabolite in die richtige Reihenfolge!: Dopamin, Adrenalin, Phenylalanin, L-DOPA, Tyrosin, Noradrenalin.
Phenylalanin -> Tyrosin -> L-DOPA -> Dopamin -> Noradrenalin -> Adrenalin
Im 1. Teil der Glycolyse entstehen aus Glucose die Konstitutionsisomere Glycerinaldehyd-3-phosphat (GADP) und Dihydroxyacetonphosphat (DHAP). Welches dieser Moleküle kann direkt und welches erst nach Isomerisierung im 2. Teil der Glycolyse weiter abgebaut werden?
GADP wird ohne Umwege abgebaut, DHAP muss zuvor zu GADP isomerisiert werden.
Welche essentielle Substanz wird für die Biosynthese von Coenzym A und die Bildung der zentralen Sulfhydrylgruppe der Fettsäuresynthase benötigt?
Pantothenat (Vitamin B5) wird für die Biosynthese von Coenzym A und 4'-Phosphopantethein benötigt. Letzteres bildet die SHz-Gruppe der Fettsäuresynthase.
Ordnen Sie die Proteine der 1. Spalte den Funktionen der 2. Spalte zu?
| Protein | Funktion |
| a) Ferritin | 1) Eisentransport |
| b) Transferrin | 2) Eisensensor |
| c) Zytosolische Isoform der mitochondrialen Aconitase | 3) Eisenaufnahme |
| d) Transferrinrezeptor | 4) Eisenspeicher |
a) Ferritin - 4) Eisenspeicher
b) Transferrin - 1) Eisentransport
c) Zytosolische Isoform der mitochondrialen Aconitase (= Eisensensorisches Protein, Iron Regulatory Protein 1, IRP1) - 2) Eisensensor
d) Transferrinrezeptor - 3) Eisenaufnahme
Wird wird Acetyl-CoA für die Fettsäurenbiosynthese von der mitochondrialen Matrix ins Zytosol transportiert?
In Form von Citrat, das in der 1. Reaktion des Citratzyklus (Kondensation von Oxalacetat mit Acetyl-CoA) gebildet wird.
Welche Funktion erfüllen Desaturasen?
Einfügung von Doppelbindungen in Fettsäuren.
Wieviel ATP liefert die vollständige Oxidation eines Glucose-Moleküls nach vorherrschender Lehrmeinung?
32
Welches Molekül ist an der Oxidation aromatischer Ringe beteiligt und woraus wird es gebildet?
Tetrahydrobiopterin (BH4) hydroxyliert Tryptophan zu Serotonin (5HT), Phenylalanin zu Tyrosin und Tyrosin zu L-DOPA. BH4 wird aus Guanosin-5'-triphosphat (GTP) gebildet
Welches Spurenelement ist für die Biosynthese von Trijothyronin und L-Thyroxin essentiell?
Iod (Iodid)
Welche 3 Aminosäuren werden für die Bildung des Tripeptids Glutathion benötigt?
Cystein, Glutamat und Glycin.
Fragen 61 - 70
BearbeitenBringen Sie die folgenden Proteine in die richtige Reihenfolge (Elektronenfluss, Benennung)!: Succinat-Dehydrogenase, Cytochrom-c-Oxidase, NADH-Dehydrogenase, ATP-Synthase, Cytochrom c, Cytochrom-c-Reduktase.
- NADH-Dehydrogenase (Komplex I)
- Succinat-Dehydrogenase (Komplex II)
- Cytochrom-c-Reduktase (Komplex III)
- Cytochrom c
- Cytochrom-c-Oxidase (Komplex IV)
- ATP-Synthase (Komplex V)
Die Umwandlung eines Alkohols zum Aldehyd und dann zur Carbonsäure entspricht chemischer einer ... ?
Oxidation (Dehydrierung). Klassisches Beispiel ist der Ethanol-Abbau.
Wo findet man folgende Reaktionssequenz? (1):
- Oxidation (Dehydrierung)
- Addition von Wasser
- Oxidation (Dehydrierung)
- Thiolytische Spaltung
- β-Oxidation der Fettsäuren
- Isoleucin-Abbau
Variationen/Teilaspekte des Themas findet man z.B. in folgenden Wegen:
- Citratzyklus (ohne 4.)
- Lysin-Abbau (zusätzliche Decarboxylierung bei 1.)
- Valin-Abbau (zusätzlich Abspaltung von CoA-SH zwischen 2. und 3. und statt 4. erfolgt eine dehydrierende Decarboxylierung)
Die Folat-abhängige Methylierung von 2'-Deoxyuridin-5'-monophosphat (dUMP) liefert ... ?
2'-Deoxythymidin-5'-monophosphat (dTMP)
Welche Aufgabe erfüllt die DOPA-Decarboxylase?
Bildung biogener Amine wie z.B. Tyramin, Dopamin, Tryptamin, Serotonin und Histamin.
Welche Aufgabe erfüllt die Monoamino-Oxidase (MAO)?
Inaktivierung biogener Amine (oxidative Desaminierung).
Welche Aufgabe erfüllt die Catechol-O-Methyltransferase (COMT)?
Die COMT ist am Abbau der Katecholamine (Dopamin, Noradrenalin, Adrenalin) beteiligt.
Welche Aufgabe erfüllt das Malat-Enzym bei der Biosynthese von Fettsäuren?
Für die Bildung von Fettsäuren wird NADPH/H+ benötigt. Wenn der NADPH/H+-Zufluss aus dem Pentosephosphatweg nicht ausreicht, dann kann mit Hilfe des Malat-Enzyms NADPH/H+ aus NADH/H+ gebildet werden. Das Malat-Enzym selbst decarboxyliert Malat zu Pyruvat und reduziert dabei NADP+ zu NADPH/H+. Das Malat entsteht bei der Reduktion von Oxalacetat mit NADH/H+.
UDP-Glucose wird für die Biosynthese von Glycogen, Glucuronsäure und Lactose benötigt. Die Bildung von UDP-Glucose erfolgt aus UTP und ... ?
Glucose-1-phosphat. Glucose-1-phosphat entsteht durch Isomerisierung von Glucose-6-phosphat oder wird beim Glycogen-Abbau freigesetzt.
Beim Katabolismus dominieren meist oxidative, beim Anabolismus reduktive Prozesse. In beiden Fällen spielen bei den Redoxreaktionen Elektronen-Carrier eine große Rolle. Welche Elektronen-Carrier sind üblicherweise an Abbaureaktionen (2) und Biosynthesen (1) beteiligt? Welche weiteren Redox-Systeme erfüllen im Körper wichtige Aufgaben? (3)
Abbau:
- NAD+ - NADH/H+
- FAD - FADH2
Aufbau:
- NADP+ - NADPH/H+
Sonstige:
- Glutathion (GSH)
- Ascorbinsäure (C) und Tocopherol (E)
- Ubichinon (Q)
- Tetrahydrobiopterin (BH4)
Fragen 71 - 80
BearbeitenÜber N- und O-glycosidische Bindungen werden Peptide/Proteine mit Glycanen verbunden. Welche Aminosäuren stellen üblicherweise die Verbindungsstellen? (1 + 2)
- N-glycosidische Bindungen: Asparagin
- O-glyosidische Bindungen: Serin und Threonin
Welche Stoffe können aus dem Terpenoid Farnesylpyrophosphat gebildet werden? (3)
- Cholesterin
- Dolicholphosphat
- Ubichinon (Coenzym Q)
Von welchem Molekül leiten sich D-Xylose und L-Iduronat ab?
D-Xylose und L-Iduronat kommen in Glycanen vor. Sie entstehen aus D-Glucuronat: L-Iduronat durch Epimerisierung, D-Xylose durch Decarboxylierung. D-Glucuronat entsteht wiederum (formal) durch Oxidation von D-Glucose.
Welches ist die wichtigste Reaktion, mit der im Körper freies Ammoniak gebunden wird?
α-Ketoglutarat + NH3 ⇔ Glutamat
Katalysiert wird die Reaktion von der Glutamatdehydrogenase (GLDH)
Welches ist das zentrale Vitamin des Aminosäurenstoffwechsels?
Pyridoxalphosphat (Vitamin B6)
Welche Aminosäuren entstehen durch nur eine Reaktion aus Intermediaten der Glycolyse bzw. des Citratzyklus?
- L-Alanin aus Pyruvat (mittels ALAT bzw. GPT, Cofaktor PALP)
- L-Aspartat aus Oxalacetat (mittels ASAT bzw. GOT, Cofaktor PALP)
- L-Glutamat aus α-Ketoglutarat (mittels GLDH)
Welche zwei nicht-essentielle Aminosäuren werde aus essentiellen Aminosäuren gebildet?
- Cystein im Methion-Abbauweg
- Tyrosin aus Phenylalanin
Welche zwei Stoffwechselwege beginnen mit der Biosynthese von Carbamoylphosphat?
- Pyridmidin-Biosynthese (Bildung von UMP)
- Harnstoffzyklus
Nennen Sie wenigstens zwei Möglichkeiten, wie die Heteropolysaccharidkette der Glycosaminoglycane nach ihrer Bildung weiter modifiziert werden kann ?
- Sulfatierung
- Epimerisierung von D-Glucuronat zu L-Iduronat
- Deacetylierung von N-Acetyl-D-Glucosamin (GlcNAc) zu D-Glucosamin (GlcN)
Welches Enzym nutzt Molybdopterin als Cofaktor?
Die Xanthin-Oxidase
Fragen 81 - 90
BearbeitenBringen Sie folgende Intermediate in die richtige Reihenfolge zur Biosynthese von Glycogen, Glucuronsäure oder Galactose:
- Glucose-6-phosphat
- UDP-Glucose
- Glucose-1-phosphat
- Glucose
Glucose -> Glucose-6-phosphat -> Glucose-1-phosphat -> UDP-Glucose
Wofür wird PRPP (α-D-5-Phosphoribosyl-1-pyrophosphat) benötigt? (3)
- Biosynthese und Recycling von Purinen
- Biosynthese von Pyrimidinen
- Biosynthese von NAD(P)+
Woher stammen die zwei Stickstoff-Atome im Harnstoff, der im Harnstoffzyklus gebildet wird?
- Carbamoylphosphat - Carbamoylphosphat wird im Mitochondrium ATP-abhängig gebildet und auf Ornithin übertragen, dabei entsteht Citrullin.
- Aspartat - Aspartatzylus: Citrullin wird unter ATP-Verbrauch mit Aspartat zu L-Argininosuccinat verbunden, das danach in Arginin und Fumarat gespalten wird.
Wieviele energiereiche Phosphorsäureanhydrid-Bindungen kostet die Bildung eines Harnstoffmoleküls im Harnstoffzyklus (ohne Berücksichtigung anhängender Reaktionen)?
4 - jeweils 2 an der Carbamoylphosphat-Synthase (2 ATP -> 2 ADP + 2 Pi) und an der Argininosuccinat-Synthase (ATP -> AMP + PPi).
Die Biosynthese von Fettsäuren am Multienzymkomplex unterscheidet sich in mehreren Punkten von der gegenläufigen β-Oxidation. Nennen Sie 3 Beispiele!
Fettsäuren-Biosynthese:
- Findet im Zytosol statt
- Verwendet Malonyl-CoA (Decarboxylierung)
- Nutzt NADPH/H+
β-Oxidation:
- Findet im Mitochondrium (Peroxisom) statt
- Liefert Acetyl-CoA
- Nutzt NAD+ bzw. FAD
Was bedeutet Substratkettenphosphorylierung? Unter welchen Umständen ist dies besonders bedeutsam?
Der Energieträger ATP (GTP) wird nicht in der Atmungskette (an der ATP-Synthase) regeneriert, sondern bei anderen Reaktionen des Intermediärstoffwechsels. Beispiele:
- Glycolyse:
- 1,3-Bisphosphoglycerat + ADP = 3-Phosphoglycerat + ATP (Phosphoglycerat-Kinase)
- Phosphoenolpyruvat + ADP = Pyruvat + ATP (Pyruvat-Kinase)
- Citratzyklus:
- Succinyl-CoA + GDP + Pi = Succinat + CoA-SH + GTP (Succinyl-CoA-Synthetase)
Bei Sauerstoffmangel z.B. im ausbelasteten Skelettmuskel ist die Substratkettenphosphorylierung in der Glycolyse die einzige Möglichkeit, die Energieversorgung der Zelle aufrechtzuerhalten.
Wie wird Stickstoff am ehesten vom Skelettmuskel zur Leber transportiert?
Über Alanin (Glucose-Alanin-Zyklus)
Was versteht man unter dem Cori-Zyklus?
Glucose-Abbau im Muskel bis zum Lactat -> Lactat-Transport zur Leber -> Dort wird aus Lactat wieder Glucose gebildet -> Glucose-Transport zum Muskel.
Wieviel NADPH/H+ liefert die vollständige Decarboxylierung eines Glucosemoleküls im HMP-Weg?
Da jede Decarboxylierung im oxidativen Teil des HMP-Weges mit der Bildung von 2 NADPH/H+ einhergeht, liefert der Abbau einer Hexose 12 NADPH/H+.
Zum Vergleich: Die Biosynthese eines Cholesterin-Moleküls aus 18 Acetyl-CoA kostet 25 Reduktionsäquivalente und 18 ATP.
Wie heißen die vier aromatischen proteinogenen Aminosäuren?
Phenylalanin, Tyrosin, Tryptophan, Histidin.
Fragen 91 - 100
BearbeitenWelche 4 typischen Symptome/Krankheitsbilder können bei Störungen der Glycolyse / Gluconeogenese beobachtet werden?
Beeinträchtigung der Glycolyse:
- Hämolytische Anämie
- Myopathie
- Neurologische Störungen
Beeinträchtigung der Gluconeogenese:
- Hypoglycämie
Literatur und Weblinks
BearbeitenWeblinks
BearbeitenDatenbanken:
- Stoffwechselwege:
- Enzyme:
- Krankheiten:
Karten:
- KEGG Pathway
- ExPASy Biochemical Pathways - Metabolic Pathways. Von Gerhard Michal.
- Maps & Charts created by Donald Nicholson
Allgemein:
- A general overview of the major metabolic pathways von Prof. Doutor Pedro Silva
- The Medical Biochemistry Page von Michael W. King, Ph.D / IU School of Medicine
- Biochemistry of Metabolism - Instructional materials for a studio-format course
- Homepage zu Stryer, „Biochemistry“ mit Molekülmodellen u. a.
- The Biochemists' Songbook MP3 Files
Literatur
Bearbeiten- Bajorat, Brandenburger: Fallbuch Biochemie. Thieme, Stuttgart (2006). ISBN 3131401915
- Berg, Tymoczko, Stryer: Biochemie. Spektrum Akademischer Verlag (2007). ISBN 3827418003
- Christen, Jaussi: Biochemie: Eine Einführung mit 40 Lerneinheiten. Springer, Berlin (2004). ISBN 3540211640
- Dettmer, Folkerts, Kächler, Sönnichsen: Intensivkurs Biochemie. Urban & Fischer Bei Elsevier (2006). ISBN 3437444506
- Garrett and Grisham: Biochemistry. 2nd ed. Volltext online
- Hofmann: Medizinische Biochemie systematisch. Uni-Med, Bremen (2006). ISBN 3895991643
- Horn, Lindenmeier, Moc, Grillhösl, Berghold, Schneider, Münster: Biochemie des Menschen. Thieme, Stuttgart (2005). ISBN 3131308834
- Horton, Moran, Scrimgeour, Perry, Rawn, Biele: Biochemie. Pearson Studium (2008). ISBN 3827373123
- Karlson, Doenecke, Koolman, Fuchs, Gerok: Karlsons Biochemie und Pathobiochemie. Thieme, Stuttgart (2005). ISBN 3133578154
- Königshoff, Brandenburger: Kurzlehrbuch Biochemie. Thieme, Stuttgart (2007). ISBN 3131364122
- Koolman, Röhm: Taschenatlas der Biochemie. Thieme, Stuttgart (2002). ISBN 3137594030
- Kremer: Crashkurs Biochemie: Repetitorium mit Einarbeitung der wichtigsten Prüfungsfakten. Urban & Fischer Bei Elsevier (2005). ISBN 3437435000
- Linnemann, Kühl, Güler: Biochemie für Mediziner: Ein Lern- und Arbeitsbuch mit klinischem Bezug. Springer, Berlin (2004). ISBN 3540211764
- Löffler, Petrides: Biochemie & Pathobiochemie. Springer-Verlag (2006). ISBN 3540326804
- Michal: Biochemie-Atlas; Biochemical Pathways, German ed. Spektrum Akademischer Verlag (1999). ISBN 3860252399
- Müller-Esterl, Brandt, Anderka, Kieß: Biochemie. Eine Einführung für Mediziner und Naturwissenschaftler. Spektrum Akademischer Verlag (2004). ISBN 3827405343
- Rassow, Hauser, Netzker, Deutzmann: Biochemie. Thieme, Stuttgart (2008). ISBN 3131253525
- Rehm, Hammar: Biochemie light. Deutsch Harri (2007). ISBN 3817118198
- Röthgens, Kreibich: Biochemie in Frage und Antwort. Urban & Fischer Bei Elsevier (2003). ISBN 3437432400
Achtung! Evtl. gibt es aktuellere Auflagen!
Autoren, Quellen und Bildnachweise
BearbeitenAutoren und Bildnachweise
BearbeitenText:
Abbildungen:
- Benutzer:NEUROtiker - Strukturformeln (Public Domain)
- User:OnkelDagobert - Abbildung Atmungskette (GNU-FDL)
- User:Rozzychan - Abbildung Atmungskette (cc-by-sa-2.5)
- User:David D. - Abbildung Malat-Aspartat-Shuttle (GNU-FDL)
- User:Gromer (PD Dr. med. habil. Stephan Gromer Shown) - Abbildung Folsäure-B12-Schnittstelle (cc-by-sa-2.5)
Der Text steht unter der Lizenz der GNU-FDL. Die Textbeiträge von Benutzer:C64 sind public domain. Die Lizenzen der Abbildungen können davon abweichen. Diese entnehmen Sie bitte den entsprechenden Bildbeschreibungsseiten auf Wikimedia Commons.
Quellen
BearbeitenOnline:
- KEGG PATHWAY Database - Pathways, Enzyme, Gene, Reaktionen, Cofaktoren
- Enzyme Structures Database (EC->PDB) - Enzyme, Reaktionen, Cofaktoren
- GNF SymAtlas - Expressionsmuster
- OMIM - Online Mendelian Inheritance in Man - Gene, Erkrankungen
- Protein Data Bank (PDB) - Molecule of the Month by David S. Goodsell - Proteine
- A general overview of the major metabolic pathways by Prof. Doutor Pedro Silva - Reaktionswege
- Wikipedia - Verschiedenes
- Fachartikel, gefunden mit PubMed (in den jeweiligen Kapiteln aufgeführt)
Print-Medien:
- Löffler, Petrides: Biochemie & Pathobiochemie. Springer-Verlag (2002). ISBN 3540422951 - Verschiedenes
Weitere Quellen und weiterführende Literatur siehe unter Literatur und Weblinks.